What is phenylketonuria
There phenylketonuria (P.K.U.) it is an autosomal recessive inherited metabolic disease that affects 1 in 10,000 individuals and appears to occur more in homozygosity than in heterozygotes.
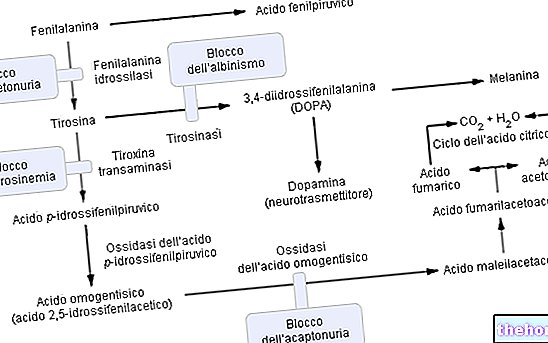
Belonging to the group of hyperphenylalaninemia, phenylketonuria significantly compromises the metabolism of phenylalanine and in particular its conversion to tyrosine; phenylketonuria is recognized by the high urinary levels of phenylalanine and some derivatives (phenylpyruvate, phenylacetate, phenylactate and phenylacetylglutamine).
The most serious complication of phenylketonuria is the mental delay.
Phenylalanine, tyrosine and derivatives
Phenylalanine is an essential amino acid and constitutes the majority of dietary proteins; it can be converted by the enzyme phenylalanine hydroxylase in tyrosine (by adding a hydroxyl group -OH). In turn, tyrosine is a precursor amino acid for the synthesis of:
- L-DOPA (dopamine synthesis intermediate)
- Epinephrine
- Norepinephrine (all neurotransmitters).

Mechanism of phenylketonuria (P.K.U.)
As anticipated, in phenylketonuria, due to one or more (6 in all) chromosomal mutations, the expression (hence the metabolic activity) of phenylalanine hydroxylase is practically nil. These alterations can be of various kinds (from "missense" changes to "splicing" defects or even "partial deletions") but what matters is that due to this enzymatic inefficiency the blood phenylalanine levels (which are normally 1mg / 100ml) in DOMINANT phenylketonuria they easily reach quantities even 50 times higher.
Functioning of the phenylalanine hydroxylase enzyme: To produce tyrosine (+ dihydrobiopterin), phenylalanine hydroxylase requires: phenylalanine, oxygen and tetrahydrobiopterin (a reduced pteridine that acts as a coofactor); the reaction is also reversible and the dihydrobiopterin can be reconverted (thanks to the enzyme dihydropterin reductase) in tetrahydrobiopterin.
Complications
Phenylketonuria can give rise to more or less severe complications based on the severity of the pathological manifestation and the timeliness of diagnosis; being a hereditary pathology, phenylketonuria is distinguished in:
- Dominant, therefore characterized by COMPLETE inactivity of the phenylalanine hydroxylase enzyme
- Recessive, in which only 30% of the total enzymatic patrimony is active.
The complications of phenylketonuria are attributable, and directly proportional, to the metabolic accumulation of phenylalanine, its derivatives and the reduced synthesis of tyrosine. In the pathology, the excess phenylalanine is filtered relatively effectively by the kidney which reabsorbs it only partially, eliminating it with urine; however, the persistence of the levels of hyper-phenylalaninemia determines a metabolic reaction of molecular CONVERSION in phenylpyruvic acid and / or other derivatives easier to drain (phenylpyruvate, phenylacetate, phenylactate).
What complicates phenylketonuria is the toxicity of phenylalanine, phenylpyruvic acid and its derivatives towards the central nervous system (CNS). their excessive presence in the brain development phase inexorably determines a form of mental retardation.
NB. The plasma concentrations of the other amino acids are slightly reduced, probably due to feedback on intestinal absorption or renal tubular reabsorption.
Brain damage, as a serious complication of phenylketonuria, is caused by the subtraction of other essential amino acids in proteosynthesis, in particular in the formation of polyribosomes, myelin, noradrenaline and serotonin. Phenylketonuria - not visible immediately after birth but after a few years - if not treated, requires hospitalization of the child and is completely irreversible.
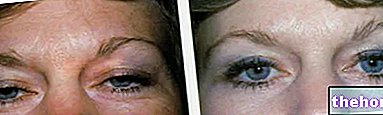
Advanced phenylketonuria can also be clearly visible to the naked eye; the high concentrations of phenylalanine, inhibiting the enzyme tyrosinase, significantly impair the synthesis of melanin by reducing skin and hair pigmentation; furthermore, the accumulation of phenylacetate in the hair and skin gives the phenylketonurics a strong and unpleasant "mouse smell".