Generality
Tuberous sclerosis is a genetic disease that affects various organs and tissues of the human body. For this reason, it presents a wide spectrum of symptoms, some typical of early childhood, others of adulthood. Tuberous sclerosis can be transmitted from parents to children, but it can also arise due to a spontaneous DNA mutation.

What is tuberous sclerosis
Tuberous sclerosis is a genetic disorder characterized by the formation of hamartomi in different organs or tissues.
Hamartoma identifies an area of tissue where cells have multiplied quite intensely, forming a noticeable mass, similar to a lump or tuber. Hamartomas are reminiscent of tumors, but they should not be confused with them: in fact, the cells of the hamartoma are identical to those of the tissue in which they proliferate; those of a tumor, on the other hand, have different characteristics. and give rise to benign neoplasms, fibroids and angiofibromas.
The brain, skin, kidneys, eyes, heart and lungs are the most affected areas, but they are not the only locations. Due to the multiplicity of organs and tissues involved, tuberous sclerosis is also defined as a multisystem genetic disease.
Later it will be understood why hamartomas appear only in certain areas.
Epidemiology
The data on the incidence and number of cases worldwide are uncertain. The uncertainty is due to the fact that many patients do not show symptoms and lead a normal life.
However, it is estimated that the incidence of tuberous sclerosis is one case for every 5,000-10,000 new births. There are about two million cases worldwide.
It causes
Tuberous sclerosis is a genetic disease; this means that a gene, present in the affected person's DNA, is mutated.
The genes that, when affected by the relative mutations, cause tuberous sclerosis are two:
- TSC1.
- TSC2.
The cases of tuberous sclerosis observed so far have only one of these genes mutated. Therefore, the single mutation of TSC1, or of TSC2, is sufficient to cause tuberous sclerosis.
Studies conducted in Europe and the United States report that the mutation in TSC2 (80% of cases) is much more frequent than in TSC1 (the remaining 20%).
TSC1 AND TSC2
The TSC1 gene resides on chromosome 9 and produces a protein called hamartin.
The TSC2 gene resides on chromosome 19 and produces a protein called tuberine.
The proteins produced, hamartin and tuberine, join and work together. This explains why the mutation of one or the other causes the same pathology.
FUNCTION OF TSC1 AND TSC2
They are considered tumor suppressor genes and play a fundamental role in the processes of:
- Cell growth and differentiation during embryogenesis.
- Protein synthesis.
- Autophagy.
When TSC1 and TSC2 are mutated, the proteins produced are defective and these physiological processes no longer take place regularly.
Cell growth and differentiation during embryogenesis
Protein synthesis
Autophagy
Cell growth and differentiation during embryogenesis
Protein synthesis
Autophagy
ONSET OF HAMARTOMAS
Hamartomas can arise when a mutation occurs in a gene that controls cell growth and differentiation, such as TSC1 or TSC2. The cells, consequently, grow in number, generating evident masses; in this way, plaques similar in shape to a nodule or a tuber are formed. In histology, this process is defined with the term hyperplasia.
GENETICS
Two premises:
- Each human DNA gene is present in two copies. Such copies are called alleles.
- The human being has 23 pairs of chromosomes. Of these, only one pair determines the sex (sex chromosomes), all the others are called autosomal chromosomes.
Tuberous sclerosis is an autosomal genetic disease dominant. For this, it is sufficient for one allele to be mutated for the entire gene to not function properly. The mutated allele, in fact, has more power than the healthy one (dominance).
In fact, tuberous sclerosis disorders are aggravated when both TSC1, or TSC2, alleles are mutated. In other words, only one allele, even if dominant over the other, does not cause evident symptoms. In these cases, we speak of alleles with incomplete dominance.
INHERITANCE € OR SPONTANEOUS MUTATION?
TSC1, or TSC2, mutation can arise from:
- Hereditary transmission (i.e. from one of the two parents) of a mutated allele.
- Spontaneous mutation of an allele in the embryonic stage (or embryogenesis).
One third of tuberous sclerosis cases are due to hereditary transmission. In these cases, it is enough that a parent has a mutation of the TSC1 or TSC2 genes for the offspring to be affected by the disease (we have in fact seen that tuberous sclerosis is an autosomal dominant hereditary disease).
The remaining 2/3 of cases are due to a spontaneous mutation during the embryonic stage.
TSC1 in 50%
TSC2 in the remaining 50%
TSC2 in 70%
TSC1 in 30%
WHY ARE ONLY CERTAIN ORGANS AFFECTED?
Premise: the embryo, during the first stages of its development, has three layers of cells:
- Ectoderm, the outermost.
- Mesoderm, the central.
- Endoderm, the innermost.
Specific organs and tissues derive from each layer.
Nervous system
Epidermis
Epithelium of the mouth
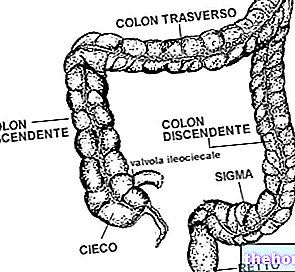
Epithelium of the colon
Horny and crystalline
Tooth enamel
Dermal bones
Heart
Kidney
Intestinal wall lining
Musculature of the limbs
Serous membranes of the lungs (pleura) and heart (pericardium).
Liver
Pancreas
Digestive system
We now have all the elements to understand why hamartomas arise only in certain parts of the body.
Mutations of TSC1 or TSC2 occur at the embryonic stage in the cells of the ectoderm and mesoderm. Therefore, the tissues, which will arise from these cell layers, will present hamartomas.
Symptoms
For further information: Tuberous Sclerosis - Causes and Symptoms
The organs and tissues affected by tuberous sclerosis are numerous. The districts most affected are:
- Brain, Skin, Kidneys, Heart, Eyes
But other, rarer ailments should not be forgotten, to the detriment of:
- Lungs, intestines, liver, teeth, endocrine system, bones
Some symptoms appear at a young age, others in adulthood.
INCOMPLETE DOMINANCE
It has already been mentioned above that the dominance of the mutated allele of the TSC1 or TSC2 genes is incomplete. This means that the healthy allele is still capable of producing a "healthy" protein (hamartin or tuberine), albeit in quantity lower. The presence of the "healthy" protein compensates for the damage caused by the mutated protein. Under these conditions, hamartomas do not yet cause dramatic manifestations.
When the other allele also changes (this is a rare event, but possible), the hamartomas grow in an uncontrolled way.
SKIN MANIFESTATIONS
About 90% of patients have skin changes. The events are numerous and varied. Typical ones are depigmented spots, Pringle's sebaceous adenomas and Koenen's nail tumors.
Depigmented spots are hypomelanotic spots, that is, with a lower melanin content
Pringle sebaceous adenomas are benign tumors also called facial angiofibromas. Hamartomas appear as small, globular, bright red masses. Koenen's nail tumors are fibroids and arise from hamartomas of a few millimeters.
Photos on the skin manifestations of tuberous sclerosis
The table shows the numerous skin manifestations due to tuberous sclerosis:
Trunk
Arts
Cheeks
Nose
Chin
Fingernails and hands
Front
Your scalp
Trunk
Dorsal-lumbar region
Neck
Shoulders
Teeth
Mouth
Anterior gum
Lip
Palate
NEUROLOGICAL SYMPTOMS
The sites of the brain affected by tuberous sclerosis are:
- The cerebral cortex
- The white matter
- The ventricles
- The basal ganglia
The two figures help the reader to understand the affected areas.

Depending on the location and shape of the hamartomas, different disorders can occur, such as:
- Epilepsy
- Subependymal nodules
- Brain tumors of the astrocytoma type
- Mental, behavioral and learning deficits.
Tuber
Bark
80-90%
- Spasms
- Partial
- Feverish
Very early childhood (spasms), 75%
Adulthood (partial), 25%
Nodule
Ventricles
80-90%
Childhood
Obstructive hydrocephalus
Evolution into subpendimal astrocytoma
Brain cysts
Nodule
> 1 cm
Ventricles (Foramina di Monro)
6%
Between 4 and 10 years
Headache
He retched
Convulsions
Alterations of the visual field
Sudden changes in mood
Hydrocephalus
Brain cysts
Mental handicap
Early childhood
(0-5 years)
Requires supervision (85%)
Absence of language (65%)
Not self-sufficient (60%)
Autism
Attention deficit
Hyperactivity
Aggression
Self-mutilation
Sleep disorders
Childhood
Association with epilepsy
Difficult family and school management
KIDNEY INJURIES
They are very frequent. In fact, they appear in 60-80% of cases. They consist of:
- Hamartomas resembling benign tumors.
- Malformations of the renal structure.
Angiomyolipoma (60-70%)
Angiolipoma
Myolipomas
They are benign tumors, which appear in multiple forms
During childhood: Asymptomatic
In adulthood: Possible rupture of the hamartoma, followed by haemorrhage, hematuria and abdominal pain.
Kidney failure
Horseshoe kidney
Polycystic kidney
Lack of a kidney (renal agenesis)
Double ureter
CARDIOVASCULAR INJURIES
Again, they are due to hamartomas similar to benign tumors, called rhabdomyomas.
Asymptomatic.
If the dimensions are large:Arrhythmias
Changes in heart flow
LUNG INJURIES
They are mainly due to pulmonary lymphangioleiomyomatosis (AML) and, to a lesser extent, to micronodular multifocal hyperplasia. They are typical manifestations of adulthood.
Rare disease
It mainly affects adult women
Lung cysts appear
Most cases are asymptomatic
Symptoms are: asthma-like dyspnoea, cough, spontaneous pneumothorax, respiratory failure
Rare disease
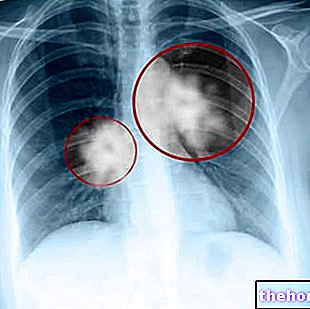
It mainly affects adults, men and women
Nodules appear, visible on a chest X-ray
Almost always asymptomatic
OTHER INJURIES
Retinal hamartoma
Retinal astrocytoma
Intestinal polyps
Intestinal cysts
Angiomyolipoma
Angiomas
Pseudo-cyst in hands and feet
Adenomas
Angiomyolipomas
Diagnosis
The diagnosis consists of:
- Anamnesis
- Clinical analysis of the aforementioned signs
- Instrumental examinations
ANAMNESIS
The doctor does a "survey on the patient's family history, to understand if tuberous sclerosis is inherited or due to a spontaneous mutation.
CLINICAL ANALYSIS OF THE SIGNS
In 1998, a group of international doctors established a diagnostic criterion based on the aforementioned clinical manifestations. They have been divided into:
- Major signs (or criteria)
- Minor signs (or criteria)
If the patient shows
- 2 major signs,
- 1 major and 2 minor signs
If the patient shows
- 1 major sign
- 2 or more minor signs
The classification of the signs is as follows:
INSTRUMENTAL EXAMINATIONS
Brain CT scan
Nuclear magnetic resonance
- Tubers of the cerebral cortex
- Subependymal nodules
- Giant cell subependymal astrocytomas (SEGA)
Yes (ionizing radiation)
No
Spirometry
Chest X-ray
- Pulmonary lymphangioleiomyomatosis
- Respiratory failure
No
Yes (ionizing radiation)
GENETIC TEST
It is a "long investigation, which takes a couple of months. It is therefore not useful for early diagnosis. Rather, it serves to confirm the diagnosis based on clinical signs."
Therapy
There is no specific and effective cure, as tuberous sclerosis is one:
- Genetic disease.
- Multisystem disease.
However, some symptoms can be curbed to avoid their complication and improve patients' quality of life.
PHARMACOLOGICAL TREATMENT
The clinical manifestations that can be treated with the administration of drugs are:
- Infantile epilepsy
- Pulmonary lymphangioleiomyomatosis (LAM)
- Kidney ailments
Infantile epilepsy. The little patient receives anti-convulsant drugs:
- ACTH (adrenocorticotropic hormone)
- Vigabatrin
Pulmonary lymphangioleiomyomatosis. Bronchodilators, of the beta-2 agonist type, such as salbutamol, are useful. However, the efficacy of hormone therapy based on progesterone or buserelin is uncertain
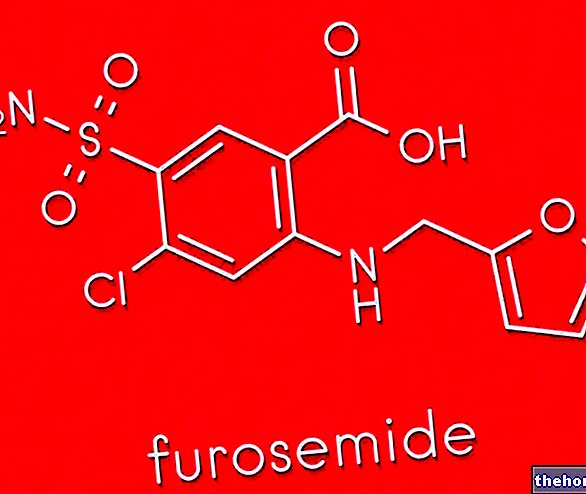
Kidney ailments. Antihypertensives, such as ACE inhibitors and diuretics, are used.
PHYSICAL-SURGICAL TREATMENTS
They consist of interventions aimed at removing:
- Facial angiofibromas
- Nail fibroids
- The skin plaques
- The knurled spots
- Giant cell subependymal astrocytomas (SEGA)
- Renal angiomyolipomas
- Lung lesions
- The tubers of the cerebral cortex, which cause epilepsy
The following table summarizes the main therapeutic treatments and their characteristics.
Diathermy
Cryotherapy
Surgical removal
Minimally invasive
Yup
Laser therapy
Surgical removal
Yup
Follow-up and prognosis
Introduction: the medical term follow-up refers to the patient who, suffering from cancer, has been positively subjected to surgery.
Periodic checks are recommended for follow-ups. Ophthalmoscopy, ie fundus examination, can also be performed once a year. Conversely, neurological, cardiac and renal conditions require more frequent monitoring.
PROGNOSIS
The evolution of tuberous sclerosis is variable and depends from case to case.
Some patients show mild, almost imperceptible symptoms. For these, the quality of life is not affected by the disease and the prognosis is excellent.
Conversely, other patients show much more dramatic and evident symptoms. Death occurs mainly due to neurological lesions, therefore, the prognosis becomes very unfavorable.
GENETIC CONSULTING
If either parent has tuberous sclerosis, the likelihood that a child will inherit the same condition is 50%.
If, on the other hand, a child of healthy parents is affected, the likelihood of a second child becoming ill is very low. In these cases, a genetic test clarifies whether the parents are carriers of tuberous sclerosis, or if, instead, a spontaneous mutation has occurred.