Generality
Mediterranean anemia (or beta-thalassemia) is an inherited blood disorder.
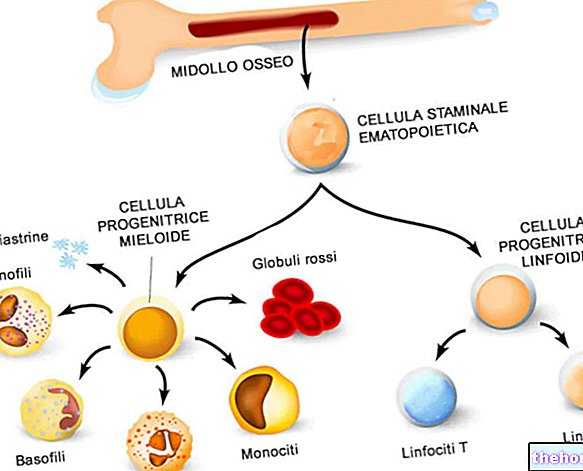
Affected patients have fewer red blood cells than normal, with defects in the synthesis of hemoglobin (Hb, the protein responsible for transporting oxygen).

The extent of the disorder, the symptoms and the consequences are very variable and fundamentally depend on the type of genetic defect. There are, in fact, 3 different forms of Mediterranean anemia:
- Thalassemia major (or Cooley's disease);
- Intermediate thalassemia;
- Minor thalassemia.
In the most serious cases, Mediterranean anemia is disabling and life-threatening; in other forms, it is almost asymptomatic. There is also the possibility of being a healthy carrier, with the risk of having children who will develop the disease.
Mediterranean anemia is detectable through genetic tests and blood tests, the latter will highlight the presence of irregularly sized, fragile, scarce and smaller than normal red blood cells.
Treatment involves several approaches, including more or less recurrent blood transfusions associated with chelation therapy (to avoid iron accumulation) and bone marrow transplantation from compatible donors. Sometimes no therapeutic intervention is necessary.















-nelle-carni-di-maiale.jpg)