Some features of Huntington's can be managed with:
- Speech therapy, psychotherapy and cognitive rehabilitation can improve both the physical and psychological symptoms of the disease. In particular, these therapies are useful for helping communication and the autonomous carrying out of daily activities. A better understanding of behavioral and cognitive disorders can also help to develop strategies to adapt to the changes induced by the progression of Huntington's disease;
- Physiotherapy and regular exercise: contribute to the maintenance of movement coordination. In the early stages of the disease, gentle physical activity (swimming, walking, etc.) is recommended;
- Use of specific aids to help Huntington's disease patients, who experience coordination difficulties, to walk independently;
- Medicines: are indicated when important symptoms occur. For example, chorea and agitation can be partially suppressed with drugs that block or deplete dopamine receptors. However, many drugs can produce side effects, as well as having different effects in different patients. Therefore, the ideal balance. of drug therapy should be established on a case-by-case basis by the specialist doctor, on the basis of symptoms and individual response to treatments.
, in the treatment of Huntington's disease. The clinical phase is very demanding, mainly because the disease has a slow progression and a "wide clinical heterogeneity. There are scales of evaluation of Huntington's disease and they are almost the same in all clinics. The complete penetrance of the disease and the availability of tests. predictive genetics, it offers the opportunity to attempt treatment during the initial stages of the disease. Currently, the studies are aimed at the search for sensitive and stable biormarkers of change, in order to intervene in the first manifestations of the disease.
Currently, neuroimaging techniques have offered the best biomarkers during the prodromal phase (which precedes the clinical symptoms of the disease); moreover, they provide a correlation between the therapies conducted on animal models and on humans.
As mentioned, striatum atrophy is early and progresses during the course of the disease. Other areas of the brain such as subcortical and cortical white matter structures have also been shown to be affected in the prodromal period.
Through functional imaging it can also identify certain abnormalities in individuals during the prodromal period. This technique may also be sensitive enough to identify detectable structure irregularities or changes in behavior.
Finally, the identification of molecular biomarkers, such as lactate or other cellular stress products, could be made possible thanks to magnetic resonance spectroscopy techniques.
the selective degeneration of neurons in Huntington's disease has not yet been fully elucidated; therefore, there is a need to investigate possible new therapeutic strategies. In particular, it has been found that in Huntington's disease there is a selective loss of CB1-type cannabinoid receptors in the basal ganglia, which represents one of the earliest neurochemical alterations. For this reason, research studies are currently investigating the neuroprotective role of cannabinoids in Huntington's disease.
For further information: Cannabinoid receptorsHuntington's disease: CB1 receptors
The involvement of the endocannabinoid system, particularly CB1 receptors, in Huntington's disease has long been hypothesized. Indeed, it has been shown that one of the first evident alterations in individuals affected by the disease is the selective loss of CB1 receptors in the basal nuclei. This receptor loss precedes the onset of striatum neuropathology. In transgenic animal models for Huntington's disease it is A change in both CB1 receptor expression and endocannabinoid levels was observed. These evidences have led to the hypothesis that a dysregulation of the endocannabinoid system could represent a target for the development of new therapeutic strategies.
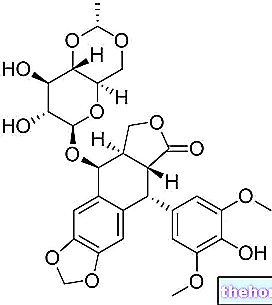
In very recent studies it has been shown that the deletion of CB1 receptors in transgenic models for Huntington's disease resulted in a worsening of the motor phenotype, in a "striatum atrophy and an accumulation of the huntingtin protein, while a chronic treatment with a cannabis agonist, tetrahydrocannibol (Δ9-THC), was beneficial.
Finally, CB1 receptors are highly expressed in GABAergic neurons, which make up 90-95% of neurons in the striatum, the brain area affected in Huntington's disease, as explained in the previous paragraphs.
Stimulation of CB1 receptors leads to a decrease in the release of the inhibitory neurotransmitter GABA. This reduction could be harmful for patients affected by Huntington's disease, given that by reducing the inhibitory tone exerted by GABA, there would be an excessive increase in the excitatory tone, determined by glutamate, and consequently to the phenomenon of excitotoxicity. Excitotoxicity is thought to contribute to the death of the projection neurons of the striatum. However, CB1 receptors are also located in glutamate neurons, albeit to a lesser extent. It has been hypothesized that stimulation of these receptors would also lead to a lower release of glutamate reducing excitotoxicity. The fact that chronic treatment with Δ9-THC was beneficial suggests that the contribution of CB1 receptors in mediating the response to cannabinoid agonists in specific neurons may change in disease progression.
Future prospects
At present, the search for a cure for Huntington's disease is active and several clinical trials are underway to evaluate the efficacy of various pharmacological agents and / or non-pharmacological approaches (eg gene therapy, stem cell transplantation) capable of reduce the production of huntingtin or improve neuronal survival, prevent or slow the progression of the disease.
For example, gene silencing using RNA interference (RNAi) or antisense oligonucleotides (ASO). The ASOs bind, in particular, to the "messenger RNA that carries information" from the mutated gene, blocks its translation and stimulates its degradation so that the huntingtin protein is not produced. Stem cell therapy, on the other hand, consists in replacing damaged neurons, thanks to the transplantation of stem cells, in the affected regions of the brain. Trials in animal models and in preliminary clinical trials have yielded conflicting results with this technique, so further evidence is needed to establish its effectiveness.
Bibliography
- Caron, N.S., Wright, G.E.B. & Hayden, M.R. Huntington Disease. GeneReviews ((R)) Huntington disease. Seattle (WA), 2018.
- Neurobiol Dis. 2012 Mar; 45: 983-91. doi: 10.1016 / j.nbd.2011.12.017. Epub 2011 Dec 23. Unbalance of CB1 receptors expressed in GABAergic and glutamatergic neurons in a transgenic mouse model of Huntington "s disease. Chiodi V, Uchigashima M, Beggiato S, Ferrante A, Armida M, Martire A, Potenza RL, Ferraro L, Tanganelli S, Watanabe M, Domenici MR, Popoli P.