Generality
Amyloidosis is the term used to define a group of diseases characterized by the accumulation, often in the extracellular area, of a fibrillar protein material, defined as amyloid. Insoluble amyloid fibrils form particularly stable deposits in numerous organs.

Other Photos Amyloidosis - Gallery 2
Symptoms and severity of the disease depend on the organ predominantly affected by amyloid accumulation and the type of amyloidosis. However, most cases are systemic. In other words, fibrillar deposits are widespread and can potentially impair function. of many tissues and organs of the body. Diagnosis is defined by biopsy, by examining a small sample of tissue under a microscope. Potential etiological factors vary according to the variant of amyloidosis. Available treatments can help manage symptoms and limit amyloid production.
Characteristics of amyloid deposits
Amyloidosis derives from disorders of the secondary structure of proteins (with β-folded sheet configuration). In normal conditions, in fact, proteins € ‹are synthesized in a linear string of amino acids, which, when folded, takes on a specific spatial conformation (protein folding). Thanks to its structure, therefore to the correct protein folding, the protein is able to perform the physiological functions for which it is responsible. The amyloid proteins derive from a precursor processed by the cells incorrectly (due to of "misfolding"). The proteins that form fibrils are diversified by size, amino acid sequence and native structure, but become insoluble aggregates that are similar in structure and properties. The precursors of fibrils are represented by primary molecules (example: light chain of immunoglobulins, β2-microglobulin, apolipoprotein A1, etc.) or from products that reflect an alteration in the s amino acid equence. The aberrant secondary structure predisposes to the formation of fibrils, which can be deposited locally in tissues and organs and lead to the impairment of their normal physiological function. More than 20 different protein precursors have been identified that can take on an amyloid conformation, which is why they exist many different types of amyloidosis.
Based on the location of the amyloid deposits, the disease can be divided into:
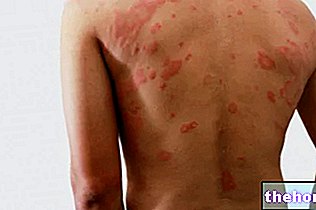
- Localized form: Confined to a particular organ or tissue (heart, kidneys, gastrointestinal tract, nervous system and dermis) and is usually less severe than systemic (diffuse) forms. For example, amyloidosis can only affect the skin, causing discoloration and / or itching. A particular type of amyloid protein has also been found in the brains of patients with Alzheimer's disease. Localized amyloidosis is typical of senescence and affected patients. from type 2 diabetes (where protein builds up in the pancreas).
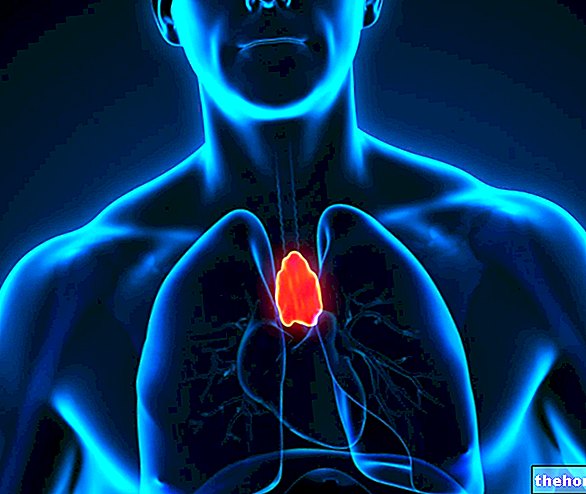
- Systemic form: amyloid deposits are present in various organs and generally recognize neoplastic, inflammatory, genetic or iatrogenic origin. Systemic amyloidosis is often very serious: it commonly damages the heart, kidneys, intestines and nerves, causing progressive insufficiency d "organ.
Classification
There are many forms of amyloidosis, classified according to the nature of the proteins that make up the fibrillar deposits.
The most common variants are:
- Primary amyloidosis (also called light chain amyloidosis, AL);
- Secondary amyloidosis (also called acquired amyloidosis, AA);
- Hereditary amyloidosis;
- Aging-associated amyloidosis (or senile systemic amyloidosis).
-cos-cause-e-disturbi-associati.jpg)