Generality
Retinoblastoma (Rb) is a malignant eye tumor that develops from the cells of the retina. This cancer can occur at any age, but onset is most common during infancy before age five.

Childhood cancer is aggressive: Retinoblastoma can spread to lymph nodes, bones, or bone marrow. Rarely, it involves the central nervous system (brain and spinal cord).
About 90% of children with retinoblastoma have a positive prognosis (probability of cure), provided that diagnosis is early and treatment is started before the cancer spreads. Whenever possible, the goal of medical intervention is to preserve the patient's vision.
Causes
The series of events leading to tumor onset is complex. This begins when cells in the retina develop a mutation (or deletion), involving the RB1 tumor suppressor gene, located on the q14 band of chromosome 13 (13q14).
Each cell normally has two RB1 genes:
- If at least one copy of the gene works correctly, retinoblastoma does not arise (but the risk increases);
- When both copies of the gene are mutated or missing, uncontrolled cell proliferation occurs.
In many cases, it is not clear what exactly induces changes in the RB1 gene (sporadic retinoblastoma); these can result from random genetic errors, which occur, for example, during reproduction and cell division. However, it is known that the genetic abnormalities underlying retinoblastoma can also be passed on from parents to children, with an autosomal dominant inheritance pattern. This means that if a parent carries a mutated (dominant) gene, each child will have a 50% chance of inheriting it and a 50% chance of having a normal genetic makeup (recessive genes).
- An occasional cell inactivates its only normal copy of the RB1 gene (one copy is already mutated);
- The loss of the two copies of RB1 leads to an "excessive proliferation of the retina.
- An occasional cell inactivates one of its normal RB1 genes;
- The second copy of the RB1 gene is inactivated;
- The loss of the two copies of RB1 induces an excessive cell proliferation which leads to retinoblastoma.
Genetic and molecular characteristics
- Retinoblastoma was the first tumor to be directly associated with a "genetic abnormality (deletion or mutation of the q14 band of chromosome 13).
- RB1 encodes the pRb protein, which plays a key role in the cell cycle: it allows DNA replication and cell cycle progression, as it participates in the transcription control of S phase genes (G1 → † "S).
- In addition to retinoblastoma, the RB1 gene is inactivated in bladder, breast and lung cancers.
Hereditary retinoblastoma
Children with hereditary retinoblastoma tend to develop the disease at an earlier age than sporadic cases. Furthermore, these children are at increased risk for other non-ocular cancers, as the abnormality in the RB1 gene is congenital (ie present from birth) and affects all cells in the body (known as a germline mutation), including those of both. retinas: For this reason, children with the hereditary form often have bilateral retinoblastoma rather than just one eye.
Symptoms
To learn more: Retinoblastoma Symptoms
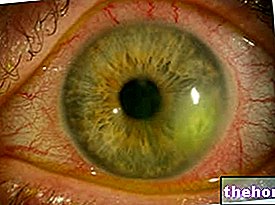
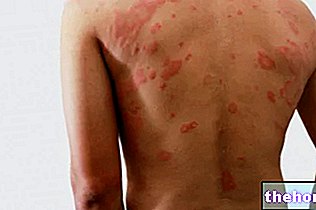
The most common and obvious sign of retinoblastoma is the abnormal appearance of the pupil, which presents a grayish-white reflection when it is hit by a beam of light (leukocoria or amaurotic cat reflex). Other signs and symptoms include: decreased vision, eye pain and redness, and developmental delay. Some children with retinoblastoma may develop a squint (misaligned eyes); in other cases, it is possible to find a neovascular glaucoma, which, after some time, can cause enlargement of the eye (buftalmo).
Cancer cells can further invade the eye and other structures:
- Intraocular retinoblastoma. Retinoblastoma can be defined intraocular when the tumor is entirely located inside the eye. The neoplasm can be found only in the retina or also affect other parts, such as the choroid, ciliary body and part of the optic nerve. Intraocular retinoblastoma, therefore, is not spread to the tissues around the outside of the eye.
- Extraocular retinoblastoma. The tumor can proliferate to affect the tissues around the eye (orbital retinoblastoma). The cancer can also spread to other areas of the body, such as the brain, spine, bone marrow, and lymph nodes (metastatic retinoblastoma).
The presence of orbital extension, uveal involvement and invasion of the optic nerve are known risk factors for the development of metastatic retinoblastoma.
Diagnosis
In case of a positive family history, the patient undergoes regular eye examinations for cancer screening. If congenital retinoblastoma is bilateral, it is usually diagnosed in the first year of life, while when it affects only one eye, the presence of the tumor can be confirmed at around 18-30 months of age.
The clinical diagnosis of retinoblastoma is established by examination of the fundus. The tumor, depending on the location, may be visible during a simple examination of the eye, through indirect ophthalmoscopy. Imaging techniques can be used to confirm the diagnosis, define the staging of the tumor (where it is, how widespread it is, whether it is affecting the functions of other organs in the body, etc.) and determine if the treatment has been effective. Investigations may include ultrasound, computed tomography (CT), and magnetic resonance imaging (MRI).
The molecular-genetic diagnosis is possible through the identification of the mutation of the RB1 gene. The cytogenetic analysis (ie of the chromosomes) of peripheral blood lymphocytes is used to detect deletions or rearrangements involving chromosome 13 (13q14.1-q14. 2).
Treatments
In case of retinoblastoma, several treatment options can be used.
The objectives of the treatment are:
- Eliminate the tumor and save the patient's life;
- Save the eye if possible;
- Preserve vision as much as possible;
- Avoid the development of other cancers, which can also be caused by treatment, especially in children with hereditary retinoblastoma.
Prognosis (probability of recovery) and treatment options depend on the following factors:
- Stage of the tumor;
- Patient's age and general health condition;
- Location, size and number of tumor foci;
- Spread of the cancer to other areas besides the eyeball
- How likely it is that vision can be preserved in one or both eyes.
Most cases of retinoblastoma are diagnosed early and successfully treated, before the cancer can metastasize outside the eyeball, resulting in a cure rate of over 90%.