Generality
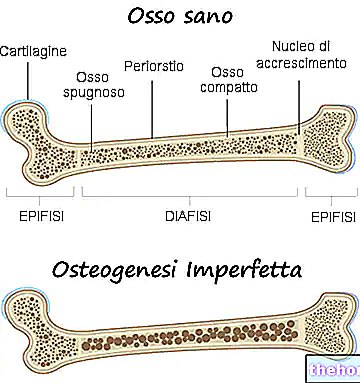
Osteogenesis imperfecta is a congenital genetic disease, not linked to sex, responsible for a certain bone fragility and a marked tendency to fractures.
The symptoms of osteogenesis imperfecta are numerous; generally, they consist of: bone weakening, high tendency to bone fractures, presence of blue, gray or purple ocular sclerae, presence of bone deformities or other skeletal alterations, triangular face, dental fragility, etc. .
In general, the following are essential for a correct diagnosis of osteogenesis imperfecta: physical examination, medical history, medical imaging tests, a type I collagen assessment test, and a genetic test.
Unfortunately, currently, the only treatments available to patients with osteogenesis imperfecta are symptomatic. The disease in question, in fact, is incurable.
What is osteogenesis imperfecta?
Osteogenesis imperfecta is a genetic disease that makes the affected person's bones weaker and more prone to fractures.
In reality, with the term osteogenesis imperfecta, doctors refer to a heterogeneous group of genetic diseases, characterized by a certain degree of bone fragility. There are, therefore, multiple forms (or types) of osteogenesis imperfecta, some much more severe than others.
IT IS A CONGENITAL DISEASE
In people who are affected by it, osteogenesis imperfecta is a disease present from birth. Therefore it can be defined, to all intents and purposes, a congenital disease.
IS IT RELATED TO SEX?
Osteogenesis imperfecta is not a gender-related genetic disease, such as haemophilia or Klinefelter's syndrome.
EPIDEMILOGY
According to some statistical research, the incidence of osteogenesis imperfecta would be equal to one case every 15,000-20,000 births. This means that every 15,000-20,000 new borns have one affected by osteogenesis imperfecta.
Other statistical studies have also shown that osteogenesis imperfecta affects males and females equally, and that it has no preference for a particular population or ethnic group.
Life span is an extremely variable parameter, which depends on the form of osteogenesis imperfecta.
Causes
Osteogenesis imperfecta almost always arises from a qualitative and quantitative alteration in the production of type I collagen.
Type I collagen is essential for strengthening bones and for maintaining healthy connective tissues constituting cartilage, tendons, skin, ocular sclera, etc.
Therefore, an alteration in the production of type I collagen affects the strength of the bones and the good health of the connective tissues present in the human body.
WHAT ALTERS COLLAGEN PRODUCTION?
A genetic disease is a condition that arises due to a mutation in one or more genes making up cellular DNA.
In the case of osteogenesis imperfecta, the causes of the latter are almost always to be found in the mutation of one or both of the genes COL1A1 (located on chromosome 17) and COL1A2 (located on chromosome 7).
Under normal conditions, COL1A1 and COL1A2 regulate the normal production of type I collagen; in the presence of mutations in their charge, they fail in their regulatory function.
Important: what other genes, if mutated, cause osteogenesis imperfecta?
In addition to the mutations of COL1A1 and COL1A2, mutations in the IFITM5, SERPINF1, CRTAP and LEPRE1 genes are potential causes of osteogenesis imperfecta.
The aforementioned genes cover functions different from COL1A1 and COL1A2 - therefore they do not control the production of type I collagen - but they still have an "influence on the strength and resistance of the bones of the human skeleton.
WHAT KIND OF GENETIC DISEASE IS IT?
Osteogenesis imperfecta is an autosomal genetic disease.
The term autosomal, associated with a genetic disease, indicates that the condition in question is due to genetic mutations based in autosomal and non-sexual chromosomes.
Readers are reminded that the human being possesses a chromosomal set of 23 pairs of total chromosomes, in which 22 pairs are of the autosomal type and only one pair are of the sexual type. The pair of chromosomes of the sexual type affects the sex of the individual .
Osteogenesis imperfecta following mutations in COL1A1, COL1A2 and IFITM5 has all the characteristics of an autosomal dominant disease. When it is due to mutations in the genes SERPINF1, CRTAP and LEPRE1, it has the characteristics of an autosomal recessive disease.
TYPES
Currently, doctors believe that there are 8 types (or forms) of osteogenesis imperfecta. To distinguish the various types, they decided to use the Roman numbering, to be precise the first eight Roman numerals.
The table below shows the 8 forms of osteogenesis imperfecta, the mutations that cause them and other characteristics.
Guy
Mutated gene
Type of genetic disease
THE
COL1A1
Autosomal dominant
II
COL1A1 and COL1A2
Autosomal dominant
III
COL1A1 and COL1A2
Autosomal dominant
IV
COL1A1 and COL1A2
Autosomal dominant
V.
IFITM5
Autosomal dominant
YOU
SERPINF1
Autosomal recessive
VII
CRTAP
Autosomal recessive
VIII
HARE 1
Autosomal recessive
* N.B: obviously, the mutations in COL1A1 and COL1A2, which cause the first four forms of osteogenesis imperfecta, are genetic alterations with slightly different characteristics. Otherwise, it would make no sense to distinguish one from the other.
Symptoms, signs and complications
All types of osteogenesis imperfecta are responsible for a weakening of the bones, such that the person affected by the disease has a particular predisposition to fractures. The degree of weakening of the bones varies according to the shape; for some of these, this weakening is greater than for others.
Having said this, it should be pointed out that each form of osteogenesis imperfecta has its own symptom picture, which for some may recall the symptom picture of other forms.
POSSIBLE SYMPTOMS AND SIGNS
The possible symptoms and signs of osteogenesis imperfecta include:
- Presence of bone malformations;
- Presence of a short and small body (intended as a trunk);
- Joint problems (ex: loose joints);
- Muscle weakness;
- Blue, purple, or gray ocular sclera;
- Triangular face;
- Barrel chest;
- Morphological anomalies of the vertebral column;
- Dental fragility;
- Decline or total loss of hearing;
- Breathing problems
- Problems related to the absence or lack of type 1 collagen.
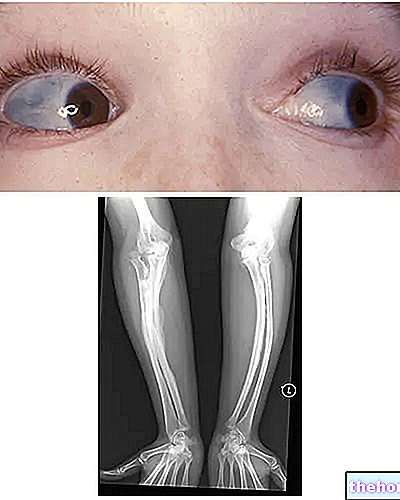
Osteogenesis Imperfecta: note the blue coloring of the sclerae and the bone deformations that characterize the disease. From wikipedia.org
WHAT ARE THE MOST SERIOUS FORMS OF IMPERFECT OSTEOGENESIS?
Doctors classify the symptomatological severity of the various types of osteogenesis imperfecta on a scale of 3 degrees, which are: the mild degree, the moderate degree and the severe degree.
Only one form belongs to the "mild degree" category: "type I osteogenesis imperfecta"; 4 forms of osteogenesis imperfecta belong to the "moderate degree" category: IV, V and VI; finally, to the "severe degree" category belong 3 forms: II, III, VII and VIII.
TYPE I: FEATURES
The most common and least severe form of all, type I osteogenesis imperfecta has the following characteristics:
- It causes fractures especially before puberty;
- It has "almost no influence on height, so patients usually have" normal height;
- Causes joint problems and muscle weakness
- It is responsible for blue, purple or gray sclera;
- It is the cause of triangular face and spinal anomalies;
- It almost never causes bone deformities. If it provokes them, they are minimal;
- It can cause dental fragility and / or hearing loss (the latter usually occurs in adulthood);
- It is associated with the presence of type I collagen that is normal in quality but abnormal in quantity (it is poorer than normal).
TYPE II: FEATURES
Type II osteogenesis imperfecta is characterized by:
- Cause of death at birth or shortly thereafter. Respiratory problems almost always cause death;
- Presence of considerable bone fragility and severe bone deformities;
- Short stature and underdeveloped lungs
- Blue, purple, or gray colored sclera;
- Presence of quantitative and qualitative anomalies of type I collagen.
TYPE III: FEATURES
Type III osteogenesis imperfecta has the following characteristics:
- Although very serious, it does not frequently cause death in the neonatal period;
- It is associated with "high bone fragility;
- It is responsible for short stature, joint problems, muscle weakness (especially in the legs and arms), barrel chest, triangular face and abnormal curvature of the spine;
- It is due to blue, purple or gray sclera;
- It can cause breathing problems, dental brittleness and hearing loss;
- It is frequently responsible for bone deformities;
- It is associated with qualitative and quantitative abnormalities of type I collagen.
TYPE IV: FEATURES
Type IV osteogenesis is characterized by:
- A degree of bone fragility between forms II and III and form I;
- Shorter than average stature;
- Blue, purple, or gray colored sclera;
- Bone deformities of mild / moderate entity, slight abnormalities of the spine and barrel chest;
- Triangular face;
- Possible presence of dental fragility and hearing loss;
- Presence of type I collagen abnormalities.
TYPE V: FEATURES
Type V osteogenesis imperfecta resembles type IV osteogenesis imperfecta in some ways. However, it has some peculiarities, which are:
- Normal colored sclera;
- Absence of dental fragility;
- Formation of abnormal bone calluses, during the healing process of fractured bones;
- Calcification of the interosseous membrane that resides between the radius and ulna. This impairs the mobility of the forearm.
TYPE VI: FEATURES
Also type VI osteogenesis imperfecta is similar to form IV. To distinguish it from the latter are some peculiarities, including the high blood levels of alkaline phosphatase and the presence, on some bones, of lamellae (bony) similar to spines of fish.
TYPE VII: FEATURES
Symptomatically, type VII osteogenesis imperfecta may resemble type IV in some circumstances and type II in other circumstances.
The peculiarities of this serious pathological form include:
- The short stature;
- The presence of an extremely short humerus (arm bone) and femur (thigh bone);
- The frequent presence of a hip deformity, known as coxa vara.
TYPE VIII: CHARACTERISTICS
Type VIII osteogenesis imperfecta is very reminiscent of forms II and III.
Among its peculiar characteristics, the following stand out: the severe growth deficit, the severe skeletal hypomineralization and the absence (or scarce presence) of the prolyl 3-hydroxylase enzyme.
Diagnosis
In general, the diagnostic process to which patients with a suspected form of osteogenesis imperfecta are subjected begins with a careful physical examination and a careful medical history; then it continues, with an "analysis of the patient's family history and with a series of diagnostic imaging tests (X-rays, CT scans, etc.); finally, it ends with a quantitative and qualitative evaluation of type I collagen and with a genetic test.
Today, there is the possibility of diagnosing osteogenesis imperfecta even in the prenatal phase, by subjecting a pregnant woman to an ultrasound.
THE IMPORTANCE OF THE OBJECTIVE EXAMINATION AND THE HISTORY
A physician experienced in osteogenesis imperfecta is able, very often, to diagnose the aforementioned disease even only by means of a physical examination and anamnesis. This means that these diagnostic tests are of no negligible importance.
EVALUATION OF TYPE I COLLAGEN PRODUCTION
As a rule, the qualitative and quantitative evaluation of type I collagen is a very reliable test, since, as stated, the majority of cases of osteogenesis imperfecta are characterized by mutations in the genes that control the production of type 1 collagen.
To assess the quantity and quality of type I collagen present at the cellular level in an individual, doctors may rely on a skin biopsy or a particular blood test.
Both of these evaluative tests are quite complex and the patient (or his parents) may have to wait several weeks to know the results.
GENETIC TEST
Through a genetic test that probes the entire DNA of the individual under examination, doctors are able to definitively decree the characteristics of the genetic mutation present.
Generally, the execution of a genetic test on all cellular DNA is foreseen when the evaluation of the characteristics of type I collagen has not provided the desired results, or when it is not a mutation in COL1A1 or COL1A2 that causes the " osteogenesis imperfecta.
PRENATAL DIAGNOSIS
Prenatal ultrasound is very useful in identifying type II and type III osteogenesis imperfecta.
Therapy
There is currently no specific cure for osteogenesis imperfecta. In other words, people with osteogenesis imperfecta are destined to live with the aforementioned condition until death, which is often due to the consequences of the disease itself.
The lack of specific therapy does not exclude the existence of other forms of treatment. In fact, among the therapeutic possibilities of a patient with osteogenesis imperfecta, various symptomatic therapies are included; by symptomatic therapies we mean treatments capable of relieving symptoms, slowing down the course of the disease and preventing (or at least postponing) the most serious consequences.
POSSIBLE SYMPTOMATIC TREATMENTS
In the list of possible symptomatic treatments for osteogenesis imperfecta, the following stand out:
- The surgical insertion, inside the longest bones (N.B: the most prone to fractures), of nails that provide greater resistance to fractures and deformities. This operation is called rodding intramedullary;
- Conservative or surgical treatment of fractures and / or bone deformities;
- Dental care, to safeguard the health of the teeth;
- Pain relieving therapies, in case of very painful multiple fractures;
- Physiotherapy, for muscle lengthening and strengthening. An elastic and tonic muscular apparatus allows you to prevent falls that could lead to various bone fractures;
- The use of aids for locomotion, including wheelchairs, braces, crutches, etc.
BENEFITS OF MOVEMENT
For individuals with osteogenesis imperfecta, doctors recommend the constant practice of physical exercise and movement in general, as both of these activities contribute to the strengthening of the skeletal and muscular system.
Among the recommended sports are: swimming, since it is a "low impact physical activity on the skeletal system", and walking.
BENEFITS FROM A HEALTHY LIFESTYLE
Leading a healthy life, avoiding smoking, drinking excess alcohol, eating too much and badly, etc., has more than discrete health benefits for patients with osteogenesis imperfecta, as it slows the progression of the disease and reduces bone fragility.
SYMPTOMATIC TREATMENTS IN THE EXPERIMENTATION PHASE
Currently, doctors and researchers are evaluating the effectiveness of some symptomatic treatments, including growth hormone treatment and bisphosphonate-based intravenous and oral therapy.
For the moment, the results provided by the aforementioned investigational treatments bode well for the entire medical community.
Prognosis
Osteogenesis imperfecta is a disease with a negative prognosis, as it is incurable, drastically compromises the quality of life and, in some cases, causes premature death of the affected subject.
However, it should be noted that, also thanks to modern symptomatic treatments, many individuals with a mild form of osteogenesis imperfecta are able to lead a pleasant and satisfying life.
Prevention
Unfortunately, there is currently no preventive measure against osteogenesis imperfecta.