Generality
Wilson's disease, also called hepatolenticular degeneration, is a rare genetic disorder characterized by an accumulation of copper in the tissues and organs of the body.

It is a fatal disease. Therefore, a therapeutic treatment is needed that removes copper from the tissues and prevents its accumulation.
So is Wilson's disease
Wilson's disease, also known as hepatolenticular degeneration, is a hereditary genetic disease that results in an excessive accumulation of copper in certain organs and tissues.
It is a rare disease, affecting 1 in every 30,000 people.
The accumulation of copper is due to a defect in its metabolism. In fact, the copper absorbed with the diet is not adequately excreted, therefore it remains in the body and is deposited mainly in:
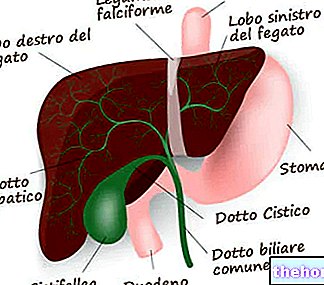
- liver;
- brain.
And to a lesser extent, also in:
- cornea;
- kidneys;
- other fabrics.
Excessive amounts of copper in these areas cause damage to the cells. The most serious effects are found in the liver and brain. In the brain, it is the lenticular nucleus that suffers the greatest consequences: hence the alternative name of hepatolenticular degeneration.
Causes
The cause of Wilson's disease is an "alteration of the ATP7B gene, located on chromosome 13, which no longer performs its normal function.
The job of the ATP7B gene is to promote the excretion, through the bile, of excess copper contained in cells. When ATP7B fails, copper builds up in such massive doses that it escapes from the cells and flows into the blood. blood, therefore, copper reaches the various tissues of the body.
Pathogenesis
Copper is consumed in the diet. Its absorption occurs in the intestine: here it binds to albumin (a plasma protein) and reaches the liver. At this point:
in a healthy individual:
- ATP7B promotes the bond between copper and ceruloplasmin. Ceruloplasmin is a plasma protein used for the transport and excretion of copper.
In an individual with Wilson's disease, however:
- ATP7B does not work. Therefore, it does not favor the bond between copper and ceruloplasmin.
- Copper remains bound to albumin, is not excreted and accumulates in liver cells.
- The liver cells saturate any copper storage capacity within them.
- The copper-albumin complex is therefore in excess. Therefore, it escapes from the hepatocytes and enters the blood.
- Through the blood, copper reaches the other tissues of the body.
The first organ to pay the consequences is therefore the liver; the brain, kidneys and cornea follow.

Why does copper spread into textiles?
In individuals with Wilson's disease, copper circulates in the blood bound to albumin. The copper-albumin bond is much more labile than that between copper and ceruloplasmin. In fact, there is little affinity between the first two c ". When the copper complexed with albumin reaches the tissues and the various organs, it encounters substances for which it has greater affinity and binds to them. The consequences are two:
- Tissues and organs are enriched with copper.
- The concentration of copper (cupremia) in the blood decreases.

From Wikipedia - Mother and father both have a mutated allele. Due to the recessive nature of this allele, they do not manifest any disease, but are healthy carriers. Both parents can transmit one mutated allele to a child each. In this case, the child will be homozygous for the given allele and will manifest the disease. In all other cases, the presence of one or both healthy alleles does not cause any disturbance.
Inheritance
Wilson's disease is an autosomal recessive inherited disease.
- Autosomal, because the ATP7B gene is located on chromosome 13, a non-sex chromosome.
- Recessive, because the mutated allele, which determines the disease, is recessive compared to the healthy one. To get sick, an individual must have both mutated alleles. In fact, a single mutated allele is not enough to cause the disease. One in 100 people circa carries an altered ATP7B allele The figure clearly explains this concept.
Symptoms
For further information: Wilson's Disease Symptoms
Although it is a hereditary genetic disease, in the very first years of age there are no disturbances. The first symptoms, based in the liver, appear around 6 years of age. This is usually the minimum time it takes for copper to accumulate in harmful quantities. In some cases, the onset can occur in late adolescence or even around the age of 30-40. Over time, disorders also appear in other tissues.
Liver symptoms
The liver is the first organ affected, because it is the first district where the copper absorbed from the diet arrives. Liver health progressively deteriorates. Evolution typically begins in adolescence and follows the following course:
- Hepatitis.
- Not severe cirrhosis.
- Severe cirrhosis.
A condition defined by the doctor is created with the term liver failure: the liver is no longer able to perform its functions.
Typical signs of liver failure are:
- Jaundice.
- Abdominal pain.
- He retched.
- Enlargement of the liver (hepatomegaly)
- Enlarged spleen (splenomegaly)
Brain symptomatology
Copper only reaches the brain when the liver can no longer keep it confined to its own cells.
Deposits in the brain cause neurological damage of a different nature:
- Physical ailments.
- Tremors of the limbs.
- Slowness of movement.
- Difficulty with speech (dysarthria).
- Difficulty in writing.
- Difficulty swallowing (dysphagia).
- Instability in walking.
- Migraine.
- Epilepsy.
- Muscle weakness and stiffness.
- Behavioral disorders.
- Mood changes.
- Depression.
- Inability to concentrate.
- Personality changes.
- Dementia.
If the patient is not treated, the neurological damage worsens more and more: the individual becomes completely dependent on others to feed and move.
Other fabrics

Furthermore, copper can also be deposited in the kidneys. Kidney damage ensues, resulting in:
- Aminoaciduria. Presence of amino acids in the urine.
- Glycosuria. Presence of glucose in the urine.
- Phosphaturia. Presence of phosphorus in the urine
- Uricosuria. Presence of uric acid in the urine.
- Calciuria. Presence of calcium in the urine.
Under normal conditions, all these lost substances would be reabsorbed. Therefore, the renal accumulation of copper alters the structure and the reabsorption of substances still useful to the organism.
Other possible symptoms of Wilson's disease are:
- Anemia.
- Pancreatitis.
- Menstrual problems.
- Miscarriage.
- Premature osteoporosis.
Diagnosis
If Wilson's disease is suspected, useful diagnostic tests are:
- Blood tests, to test:
- Concentrations of ceruloplasmin. A low level, below 20mg / 100ml, is indicative of the disease. The normal value is 30 mg / 100ml.
- The concentration of copper (cupremia). If it is less than normal, it is indicative of the disease.
- Possible haemolytic anemia.
- Liver and kidney functions, through their respective markers (transaminases, azotemia, etc.)
- Urinalysis, to evaluate the amount of copper present (cupruria). Levels above normal are indicative of the disease. Typically, people with the condition excrete about 100μg of copper in their urine every 24 hours.
- Optometric examination, to detect the presence of the Kaiser-Fleischer ring.
- Liver biopsy, to measure the copper content in liver cells. The pathological level of copper is over 100μg per gram of liver. It is also useful for assessing the state of cirrhosis.
- An MRI of the brain, to evaluate the health of the lenticular nucleus, which we remember is the area of the brain affected by the accumulation of copper.
- A genetic DNA test.
The simultaneous presence of:
- Kaiser-Fleischer ring.
- Signs of liver cirrhosis.
- Lesion of the lenticular nucleus.
leave no doubt about the diagnosis.
Measured parameter
Cupremia
110 μg / ml
<100μg / ml
Cupruria
100 μg / 24h
>> 100 μg / 24h
Ceruloplasmin
30 mg / ml
<20 mg / ml
Therapy
See also: Drugs for Adrenal Insufficiency
Left untreated, Wilson's disease is fatal. Death can occur even a couple of years after the first symptoms appear. The patient is subject to a progressive worsening of his condition, becomes increasingly dependent on others, and in the absence of specific treatment, liver and brain damage can be irreversible.
Therapy consists of:
- Reduce copper deposits in the liver.
- Control the absorption of copper in the intestine.
- Reduce the introduction of copper taken with the diet.
- Liver transplantation.
Reduce copper deposits
It is the most important step in saving the patient's life. It is based on the administration of:
- Penicillamine.
- Trientina.
Penicillamine is the drug of choice. Its administration takes place orally and must be taken for life. It represents a chelating agent capable of sequestering excess copper and conducting it to the kidneys for excretion. However, it can cause undesirable effects in the kidney itself. In these cases, it is advisable to interrupt the treatment to resolve the problems that have arisen, and to adopt an alternative based on trientine.
Trientine is also a chelating agent. It is administered orally and acts like penicillamine. It is not as effective, but the side effects are also less.
Check intestinal absorption of copper
It is possible to reduce the absorption of copper by taking zinc. This prevents the accumulation of copper in the liver. Administration of zinc is recommended when Wilson's disease is in its early stages. In other words, when the copper has not yet invaded the other tissues. Therapy is effective when combined with penicillamine treatment.
Reduce the introduction of copper
The consumption of certain foods rich in copper should be limited, such as:
- Walnuts.
- Liver.
- Mushrooms.
- Chocolate.
- Seafood.
Overall, the daily dietary intake of copper should not exceed 2 mg.
Liver transplantation
This is the therapy required if:
- Liver damage is irreversible. In this case, we speak of severe cirrhosis.
- Previous treatments have been ineffective.
Prognosis
The earlier therapy is initiated, the better the prognosis and quality of life will be.
Intervening late means limiting and only partially improving liver and brain damage due to excess copper. Some functions, in fact, remain irreparably compromised.
In severe cases, the only solution for a better prognosis is a liver transplant.