To talk about the twenty amino acids that make up the protein structures and the modified ones, it would be necessary to describe at least twelve specialized metabolic pathways.
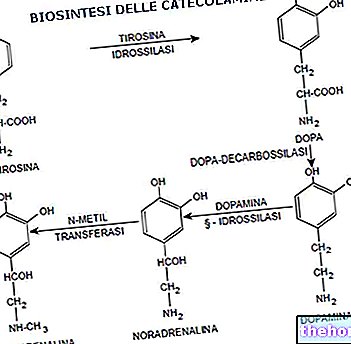
But why do cells use so many metabolic pathways that require energy (for example to regenerate the catalytic sites of enzymes), each with an enzymatic patrimony, to catabolize amino acids? From almost all amino acids it is possible to obtain, through specialized pathways, metabolites which are in small part used to produce energy (for example, through gluconeogenesis and the pathway of ketone bodies) but which, above all, lead to the formation of complex molecules, with a high number of carbon atoms (for example from phenylalanine and tyrosine, hormones are produced in the adrenal glands which are specialized for this purpose); if on the one hand it would be simple to produce energy from amino acids, on the other it would be complicated to build complex molecules starting from small molecules: the catabolism of amino acids allows them to exploit their skeleton to obtain larger species.
Two or three hectograms of amino acids are degraded daily by a healthy individual: 60-100 g of them derive from the proteins introduced with the diet but over 2 hectograms are obtained from the normal turnover of proteins that are an integral part of the organism (amino acids of these proteins, which are damaged by redox processes, are replaced by others and catabolized).
Amino acids give an energy supply in terms of ATP: after removing the α-amino group, the remaining carbonaceous skeleton of amino acids, following suitable transformations, can enter the krebs cycle. Furthermore, when the supply of nutrients is deficient and the quantity of glucose decreases, gluconeogenesis is activated: gluconeogenetic amino acids are called those which, after appropriate modifications, can be introduced into gluconeogenesis; gluconeogenetic amino acids are those that can be converted into pyruvate or in fumarate (fumarate can be converted into malate that leaves the mitochondrion and, in the cytoplasm, is transformed into oxaloacetate from which phosphoenol pyruvate can be obtained). vinegar-acetate.
The one just described is a very important aspect because amino acids can remedy a sugar deficiency in case of immediate fasting; if fasting persists, after two days the lipid metabolism intervenes (because the protein structures cannot be attacked too much) it is in this phase that, since gluconeogenesis is very limited, the fatty acids are converted into acetyl coenzyme A and ketone bodies . From further fasting, the brain also adapts to use the ketone bodies.
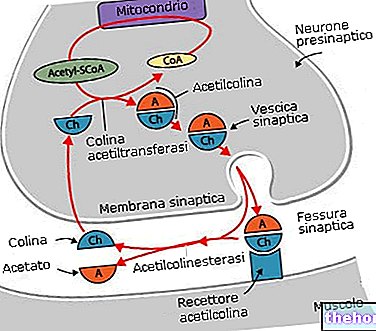
The transfer of the α-amino group from amino acids occurs through a transamination reaction; the enzymes that catalyze this reaction, they say, in fact, transaminases (or amino transferase). These enzymes use an enzymatic cofactor called pyridoxal phosphate, which intervenes with its aldehyde group. Pyridoxal phosphate is the product of the phosphorylation of pyridoxine which is a vitamin (B6) found mainly in vegetables.
Transaminases have the following properties:
High specificity for a ketoglutarate-glutamate α pair;
They are named after the second couple.
Transaminase enzymes always involve the α ketoglutarate-glutamate pair and are distinguished according to the second pair involved.
Examples:
L"aspartate transaminase ie GOT (Glutamate-Ossal acetate Transaminase): the enzyme transfers the α-amino group from "aspartate to" α-ketoglutarate, obtaining oxaloacetate and glutamate.
L"alanine transaminase ie GTP (Glutamate-Pyruvate Transaminase): the enzyme transfers the α-amino group from "alanine to" α-ketoglutarate, obtaining pyruvate and glutamate.
The various transaminases use α-ketoglurate as an acceptor of the amino group of amino acids and convert it into glutamate; while, the amino acids that are formed are used in the pathway of the ketone bodies.
This type of reaction can happen in both directions since they break and form bonds with the same energy content.
The transaminases are both in the cytoplasm and in the mitochondrion (they are mostly active in the cytoplasm) and differ in their isoelectric point.
The transaminases are also able to decarboxylate amino acids.
There will have to be a way to convert glutamate back to α-ketoglutarate: this is done by deamination.
There glutamate dehydrogenase it is an enzyme capable of transforming glutamate into α-ketoglutarate and, therefore, of converting the amino groups of amino acids found in the form of glutamate into ammonia. What takes place is a redox process that passes through the intermediate α-amino glutarate: ammonia and α-ketoglutarate are released and return to the circulation.
Then, the disposal of the amino groups of the amino acids passes through the transaminases (which differ according to the substrate) and the glutamate dehydrogenase, which determines the formation of ammonia.
There are two types of glutamate dehydrogenase: cytoplasmic and mitochondrial; the cofactor, which is also the cosubstrate of this enzyme is NAD (P) +: glutamate dehydrogenase uses either NAD + or NADP + as an acceptor of reducing power. The cytoplasmic form prefers, although not exclusively, NADP + while the mitochondrial form prefers NAD +. The mitochondrial form has the purpose of disposing of amino groups: it leads to the formation of ammonia (which is a substrate for a specialized enzyme in the mitochondrion) and NADH (which is sent to the respiratory chain). The cytoplasmic form works in the opposite direction i.e. it uses ammonia and α-ketoglutarate to give glutamate (which has a biosynthetic destination): this reaction is a reductive biosynthesis and the cofactor used is NADPH.
Glutamate dehydrogenase works when it is necessary to dispose of the amino groups of amino acids such as ammonia (via urine) or when the skeletons of amino acids are needed to produce energy: this enzyme will therefore have as negative modulators the systems that are an indication of good energy availability (ATP, GTP and NAD (P) H) and as positive modulators, the systems that indicate a need for energy (AMP, ADP, GDP, NAD (P) +, amino acids and thyroid hormones).
Amino acids (mainly leucine) are positive modulators of glutamate dehydrogenase: if amino acids are present in the cytoplasm, they can be used for protein synthesis, or they must be disposed of because they cannot be accumulated (this explains why amino acids are positive modulators) .
Disposal of ammonia: urea cycle
Fish dispose of ammonia by introducing it into water through the gills; birds convert it into uric acid (which is a condensation product) and eliminate it with feces. Let's see what happens in humans: we have said that glutamate dehydrogenase converts glutamate in α-ketoglutarate and ammonia but we have not said that this occurs only in the mitochondria of the liver.
A fundamental role of ammonia disposal, through the urea cycle, is played by mitochondrial transaminases.

Carbon dioxide, in the form of bicarbonate ion (HCO3-), is activated by the biotin cofactor forming carboxy biotin which reacts with ammonia to give carbamic acid; the next reaction uses ATP to transfer a phosphate onto the carbamic acid forming carbamyl phosphate and ADP (the conversion of ATP into ADP is the driving force for obtaining carboxybiotine). This phase is catalyzed by carbamyl phosphate synthetase and occurs in the mitochondrion. Carbamyl phosphate and ornithine are substrates for the enzyme ornithine trans carbamylase which converts them to citrulline; this reaction occurs in the mitochondria (hepatocytes). The citrulline produced leaves the mitochondrion and, in the cytoplasm, goes under the "action of"arginine succinate synthetase: there is the fusion between the carbonaceous skeleton of citrulline and that of an aspartate through a nucleophilic attack and subsequent elimination of water. The arginine succinate synthetase enzyme requires an ATP molecule so there is an energetic coupling: the hydrolysis of the ATP to AMP and pyrophosphate (the latter is then converted into two molecules of orthophosphate) occurs by the expulsion of a molecule d "water from the substrate and not by the action of the water of the medium.
The "next enzyme is the"arginine succinase: this enzyme is able to split arginine succinate into arginine and fumarate within the cytoplasm.
The urea cycle is completed by the enzyme arginase: urea and ornithine are obtained; urea is disposed of by the kidneys (urine) while ornithine returns to the mitochondrion and resumes the cycle.
The urea cycle is subject to indirect modulation by arginine: the accumulation of arginine indicates that the urea cycle must be speeded up; the modulation of arginine is indirect because arginine positively modulates the acetyl glutamate synthetase enzyme. The latter is able to transfer an acetyl group on the nitrogen of a glutamate: N-acetyl glutamate is formed which is a direct modulator of the carbamyl-phospho synthetase enzyme.
Arginine accumulates as a metabolite of the urea cycle if the production of carbamyl phosphate is not sufficient to dispose of the ornithine.
Urea is produced only in the liver but there are other sites where the initial reactions take place.
The brain and muscles use special strategies to eliminate amino groups. The brain uses a very efficient method in which an enzyme is used glutamine synthetase and an enzyme glutamase: the first is present in neurons, while the second is found in the liver. This mechanism is very efficient for two reasons:
Two amino groups are transported from the brain to the liver in a single vehicle;
Glutamine is much less toxic than glutamate (glutamate also carries out neuronal transfer and must not exceed the physiological concentration).
In fish, a similar mechanism brings the amino group of amino acids to the gills.
From the muscle (skeletal and cardiac), the amino groups reach the liver through the glucose-alanine cycle; the enzyme involved is the glutamine-pyruvate transaminase: it allows the transposition of amino groups (which are in the form of glutamate), converting pyruvate into alanine and, at the same time, glutamate into α-ketoglutarate in the muscle and, catalyzing the reverse process in the liver.
Transaminases with different tasks or positions also have structural differences and are determinable by electrophoresis (they have different isoelectric points).
The presence of transaminases in the blood may be a symptom of liver or heart damage (ie tissue damage to liver or heart cells); the transaminases are in very high concentrations both in the liver and in the heart: through electrophoresis it is possible to establish whether the damage has occurred in the liver or heart cells.




.jpg)