Definition of thalassemia
Thalassemia is a genetically transmitted blood disorder in which the body synthesizes an abnormal form of hemoglobin.
As most people know, hemoglobin is a protein contained in red blood cells, essential for the transport of oxygen in the blood. In subjects suffering from thalassemia, the mutated form of hemoglobin causes the gradual but inexorable destruction of red blood cells, up to anemia.
From the medical statistics it is clear that thalassemia mainly affects the inhabitants of Middle Eastern countries, African countries and all those who populate swampy places (not surprisingly, thalassemia is also called Mediterranean anemia).
Classification and causes
According to the defective protein sub-unit (which makes up hemoglobin), two forms of thalassemia are distinguished: before proceeding with the analysis, let's take a step back to clarify some very important concepts.
Hemoglobin is the carrier par excellence, used to transport oxygen in the blood; it is made up of two proteins, known as alpha-globulin and beta-globulin.
Thalassemia occurs when one or more genes that control the production of one or both of these proteins are defective (mutated).
Thalassemia is caused by a DNA mutation of the proteins that make up hemoglobin: these alterations have a heavy impact on the physiological synthesis of hemoglobin and, by destroying the erythrocytes, lead to anemia.
The classification of thalassemia must be made on the basis of two important factors:
- Number of mutated genes inherited from parents
- Type of protein involved (alpha or beta hemoglobin)
Alpha Thalassemia
In the "alpha" form of thalassemia - in which the 4 "alpha" globular subunits of hemoglobin (at chromosome 16) can be mutated - one or more defective genes are involved; each globular subunit is clearly encoded , from a gene, therefore the genes that can be involved are 4.

The general symptom picture becomes more serious when three or four genes are involved: in the first case, we speak of "hemoglobin H disease"(With moderate or severe symptoms). When all four genes are involved, the disease is called alpha-thalassemia major: in similar situations, the newborn dies shortly before birth or soon after.
Beta Thalassemia
The beta form of thalassemia, as can be guessed, occurs when the genes involved in the composition of the beta chains are mutated (at the level of chromosome 11): in this case, only two genes can be affected. If only one gene is changed, it is referred to as beta-thalassemia minor, in which the patient does not complain of relevant symptoms. Similarly to the alpha variant, the involvement of both genes making up the beta chains of hemoglobin results in one beta-thalassemia major (or Cooley's anemia), which reflects severe and severe symptoms; in this case, however, the symptoms usually begin after a couple of years from birth.
Watch the video
- Watch the video on youtube
Symptoms
For further information: Thalassemia Symptoms
Thalassemia is a very serious hereditary disease, so much so that some of its variants, such as alpha-thalassemia major, can cause the baby to die during delivery or soon after birth. Infants with beta-thalassemia major, however, can survive and develop the first symptoms within a couple of years of birth (severe anemia).
If only one gene is altered, both in the alpha and in the beta form of thalassemia, patients do not complain of any appreciable symptoms; only through the microscope analysis of a blood sample taken from the patient is there an abnormality in the shape and structure of the erythrocytes, much smaller than the norm.
In addition to anemia, patients with thalassemia may experience one or more of the following symptoms: fatigue, mood changes (irritability), growth failure, facial bone deformities, jaundice, shortness of breath, and dark urine.
In cases of severity, the symptomatological picture of a patient suffering from thalassemia can degenerate, to the point of creating real bone deformities, especially on the face and skull; thalassemia can promote an "abnormal expansion of the bone marrow, both by making the bone mass fragile and by enormously increasing the risk of bone fractures.
Among the complications of thalassemia, the possible accumulation of iron (hemochromatosis), an expression both of the disease itself and of the recurrent blood transfusions that the patient needs, should also be mentioned.
Thalassemia often causes splenomegaly, that is to say an exaggerated volumetric increase in the spleen: often, this pathological clinical condition requires splenectomy, the surgical removal of the organ. As we know, the spleen is an important organ used for the synthesis of blood cells and antibodies, in addition to infection control: its removal, clearly, favors a reduction in the defense function against bacterial and viral insults, making the subject more sensitive to infections. However, it should be pointed out that thalassemia itself also increases the risk of contract infections: in case of excision of the spleen in the context of thalassemia, the chances of infection increase exaggeratedly.

Diagnosis
If the father and / or mother are affected by thalassemia, the probability of transmitting the disease to the offspring is very high. We have analyzed that not all forms of thalassemia begin with a precise symptomatology right from birth: in similar situations, in case of suspected thalassemia, it is possible to subject the patient to a series of specific tests and examinations, aimed at the diagnostic assessment (such as determination of hemoglobin A2, which is elevated in healthy subjects carrying Beta-thalassemic genes).
Among the physical examinations, medical palpation of the spleen can sometimes ascertain a thalassemia: splenomegaly, as previously mentioned, constitutes a first alarm signal for Mediterranean anemia. Blood tests are more specific and precise: in a blood sample from a thalassemia, the red blood cells, when viewed under a microscope, appear small and have an abnormal shape. Furthermore, a careful blood count of a patient suffering from thalassemia reveals severe anemia: this test is useful for the iron count in the blood, to perform the DNA analysis for the diagnostic assessment of the disease and to evaluate the possible mutation of the "hemoglobin.
On the other hand, electrophoresis of hemoglobins reveals the abnormal shape of the oxygen-carrying proteins.
Some variants of thalassemia cannot be diagnosed with electrophoresis: in this case, the patient will be subjected to a "mutational analysis" test, which is useful for detecting and ascertaining thalassemia.
Medicines and treatments
See also: Medicines for the treatment of thalassemia
Being a genetically transmitted disease, it is understandable that - for the moment - there is no drug capable of reversing the disease; however, it is possible to control the symptoms, improving the patient's quality of life. The choice of one treatment rather than another depends on the type of thalassemia and the severity of the symptoms.
In the mild variant of thalassemia (in which, for example, only one gene is mutated) no drugs are needed, since the patient does not complain of symptoms. In such circumstances, it is still advisable to carry out the necessary checks regularly; Occasionally, occasional blood transfusions are sometimes useful (especially in the case of surgery and childbirth).
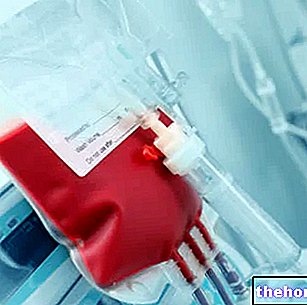
For moderate or severe symptomatic forms, the treatment approach is different, and may require frequent blood transfusions or, in severe cases, stem cell transplantation.
- Blood transfusions: this therapeutic approach can also create serious complications, since frequent transfusions can favor a pathological accumulation of iron in the blood (hemochromatosis), which requires specific treatment aimed at eliminating iron storage, known as therapy chelator (with drugs such as Deferasirox and Deferiprone). For further information: read the article on drugs for the treatment of hemochromatosis.
- Bone marrow transplant: reserved for the most serious cases, in which thalassemia creates severe dysfunctions in the body.
-cosa-significa-quando-preoccuparsi.jpg)