Active ingredients: Risedronic acid (Risedronate sodium)
MEDEOROS 35 mg film-coated tablets
Indications Why is Medeoros used? What is it for?
MEDEOROS belongs to a group of non-hormonal medicines called bisphosphonates which are used to treat bone diseases (osteoporosis). It acts directly on the bones, strengthening them and thus reducing the risk of fractures.
Bone is living tissue. The body continually removes old bone tissue and replaces it with new bone.
Postmenopausal osteoporosis is a condition that develops in women after the menopause when there is a weakening and thinning of the bones with a consequent increased risk of fractures following falls or strain.
Osteoporosis can also occur in men from various causes such as aging and / or a low level of the male hormone, testosterone.
The bones most prone to fracture are those of the spine, hip and wrist, although all bones in the body can fracture. Fractures associated with osteoporosis can also cause back pain, loss of height (weight loss). , sagging of the back (hump). Many patients with osteoporosis have no symptoms and don't even know they have it.
MEDEOROS is used for the treatment of osteoporosis:
- in postmenopausal women even in case of severe osteoporosis. Reduces the risk of fractures of the vertebrae and hip
- in men at high risk of fractures.
Contraindications When Medeoros should not be used
Do not take MEDEOROS:
- if you are allergic to risedronate sodium or any of the other ingredients of this medicine;
- if you suffer from a condition called hypocalcaemia (a low level of calcium in the blood);
- if you are pregnant, suspect or are planning to become pregnant;
- if you are breast-feeding
- if you have severe kidney problems.
Precautions for use What you need to know before taking Medeoros
Talk to your doctor or pharmacist before taking MEDEOROS In particular:
- If you have ever had problems with your esophagus (the tube that connects your mouth to your stomach) which caused pain or difficulty in swallowing food
- If you are unable to keep your upper body straight (sitting or standing) for at least 30 minutes from the time you take the tablet;
- If you have or have recently had problems with your esophagus, including Barrett's esophagus (a condition associated with changes in the cells that line the lower esophagus);
- If you have disturbances in bone and mineral metabolism (eg vitamin D deficiency, parathyroid hormone dysfunction which both lead to a reduction in blood calcium levels).
- If you have or have had pain, swelling or numbness in the jaw or a 'heavy jaw feeling' or looseness of a tooth.
- If you are being treated by your dentist or planning a dental operation, please inform your dentist that you are being treated with risedronate sodium.
- If you suffer from an "intolerance to some sugars (such as lactose, milk sugar). Patients with rare hereditary problems of galactose intolerance, lactase deficiency or glucose-galactose malabsorption should not take this medicine. Your doctor will tell you what to do. to do while taking MEDEOROS if you have any of the above conditions.
Children and adolescents
The use of risedronate sodium is not recommended in children and adolescents below 18 years of age as data on safety and efficacy are insufficient.
Interactions Which drugs or foods can change the effect of Medeoros
Medicines containing one of the following components decrease the effect of MEDEOROS when taken at the same time:
- football
- magnesium
- iron
- aluminum (for example some mixtures for digestive difficulties)
Take these medicines at least 30 minutes after taking MEDEOROS.
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines.
MEDEOROS with food and drink
It is very important that you DO NOT take food or drinks (except tap water) together with your MEDEOROS tablet so that it works properly. In particular, do not take this medicine at the same time as dairy products (such as milk) as they contain calcium ( see section 2 "Other medicines and MEDEOROS").
Take food and drink (except tap water) at least 30 minutes after the MEDEOROS tablet.
Warnings It is important to know that:
Pregnancy and breastfeeding
DO NOT take MEDEOROS if you are pregnant, think you may be pregnant or are planning to become pregnant (see section 2 "Do not take MEDEOROS"). The potential risk associated with the use of risedronate sodium (active ingredient of MEDEOROS) in pregnant women is not known.
DO NOT take MEDEOROS if you are breast-feeding (see section 2 "Do not take MEDEOROS").
MEDEOROS is only to be used for the treatment of postmenopausal women and men.
Driving and using machines
No effect on the ability to drive and use machines was observed.
MEDEOROS contains lactose
If you have been told by your doctor that you are intolerant to some sugars, contact your doctor before taking this medicine (see section 2 "Warnings and precautions").
Dose, Method and Time of Administration How to use Medeoros: Posology
Always take this medicine exactly as your doctor has told you. If in doubt, consult your doctor or pharmacist.
The recommended dose is 1 MEDEOROS tablet (35 mg risedronate sodium) once a week. Choose the day of the week that best fits your activities. Take one MEDEOROS tablet once a week on your chosen day.
The box has some compartments / spaces. Write down the day of the week you have chosen to take your MEDEOROS tablet. Note also the days on which you will take the tablet.
Take the tablet at least 30 minutes before your first meal of the day, your first drink, other than tap water, or before other medicines.
Take the tablet while standing upright (sitting or standing) to avoid heartburn. Swallow the tablet with at least one glass of tap water (120 ml). The tablet should be swallowed whole. Do not chew or let the tablet melt in your mouth. Do not lie down for 30 minutes after swallowing the tablet.
Your doctor will tell you if you need calcium and vitamin supplements if they are not getting enough in your diet.
If you forget to take MEDEOROS
If you forget to take your tablet at the usual time, take it on the day you remember.
Resume taking one tablet once a week on the day you originally chose.
DO NOT take two tablets on the same day to make up for a forgotten tablet.
If you stop taking MEDEOROS
If you stop taking MEDEOROS you may start to lose bone mass. Talk to your doctor before deciding to stop taking this drug.
If you have any further questions on the use of this medicine, ask your doctor or pharmacist.
Overdose What to do if you have taken too much Medeoros
If you or someone else have accidentally taken more MEDEOROS tablets than prescribed, drink a full glass of milk and consult your doctor immediately.
Side Effects What are the side effects of Medeoros
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Stop taking MEDEOROS and contact a doctor immediately if you experience any of the following side effects:
- Symptoms of a severe allergic reaction, such as:
- swelling of the face, tongue or throat
- difficulty swallowing
- wheals (raised, red patches of skin) and difficulty in breathing
- Severe blistering skin reactions, including blistering.
Tell your doctor promptly if you notice the following side effects: Inflammation of the eyes, usually with pain, redness and sensitivity to light.
Necrosis (destruction) of the bone in the jaw (osteonecrosis) associated with delayed healing and the onset of infection, often following tooth extraction (see section 2 "Before taking MEDEOROS").
Disorders of the esophagus such as pain in swallowing, difficulty in swallowing, chest pain or onset / worsening of heartburn.
However, the other side effects observed in clinical trials were usually mild in nature and did not require patients to discontinue treatment.
Common (affects 1 to 10 users in 100):
- Dyspepsia, feeling nauseous, stomach pain, stomach cramps or stomach discomfort, constipation, bloating, bloating (increased intestinal air), diarrhea.
- Pain in the bones, muscles or joints.
- Headache.
Uncommon (affects 1 to 10 users in 1000):
- Inflammation or ulcer of the esophagus (the tube that connects the mouth to the stomach) which causes difficulty and pain in swallowing (see section 2 "Before taking MEDEOROS"), inflammation of the stomach and duodenum (the first portion of the intestine that follows stomach). - Inflammation of the colored part of the eye (iris) (painful red eyes with possible impaired vision).
Rare (affects 1 to 10 users in 10,000):
- Inflammation of the tongue (swollen red and sometimes painful), narrowing of the esophagus (the tube that connects the mouth to the stomach).
- Abnormalities in liver function tests have been reported. These can be diagnosed by a blood test.
The following undesirable effects have been reported during post-marketing experience:
Not known (frequency cannot be estimated from the available data)
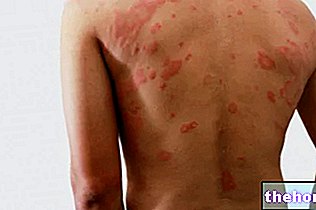
- Allergic skin reactions such as hives (hives), skin rash (sudden redness of the skin), swelling of the face, lips, tongue and / or neck, difficulty in swallowing or breathing;
- Severe skin reactions including blistering under the skin; inflammation of small blood vessels, characterized by palpable red spots on the skin (leukocytoclastic vasculitis);
- A serious condition called Stevens Johnson syndrome (SJS) with blisters on the skin, mouth, eyes and other moist areas of the body (genitals); a serious disease called toxic epidermal necrolysis (TEN) which causes a red rash on many parts of the body and / or peeling of the outer skin layers.
- Hair loss
- Allergic reactions (hypersensitivity).
- Severe liver problems, especially if you are being treated with other medicines known to cause liver problems.
- Inflammation of the eye causing pain and redness.
Rarely at the start of treatment, the patient's blood calcium and phosphate levels may decrease.
These changes are usually mild and asymptomatic.
Rarely, an unusual fracture of the femur may occur particularly in patients on long-term treatment for osteoporosis. Contact your doctor if you experience pain, weakness or discomfort in the thigh, hip or groin as this may be an early indication. of a possible fracture of the femur.
Very rare (affects up to 1 in 10,000 people)
- Talk to your doctor if you have ear pain, ear discharge and / or ear infection. These episodes could be signs of bone damage in your ear.
Reporting of side effects
If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. You can also report side effects directly via the Italian Medicines Agency website: www.agenziafarmaco.gov.it/it/responsabili. By reporting side effects you can help provide more information on the safety of this medicine.
Expiry and Retention
Keep this medicine out of the sight and reach of children.
This medicine does not require any special storage conditions.
Do not use this medicine after the expiry date which is stated on the carton. The expiry date refers to the last day of that month.
Do not throw any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. This will help protect the environment.
Other information
What MEDEOROS contains
- The active ingredient is risedronate sodium. Each tablet contains 35 mg of risedronate sodium (as risedronate sodium hemipentahydrate).
- The other ingredients are: Core: Microcrystalline cellulose, crospovidone, magnesium stearate, lactose monohydrate.
- Coating: red iron oxide, yellow iron oxide, anhydrous colloidal silica, titanium dioxide, macrogol 400, macrogol 8000, hypromellose, hydroxypropyl cellulose.
Description of what MEDEOROS looks like and contents of the pack
MEDEOROS are round, light orange film-coated tablets 9 mm in diameter.
They are available in blisters containing 4 tablets.
Source Package Leaflet: AIFA (Italian Medicines Agency). Content published in January 2016. The information present may not be up-to-date.
To have access to the most up-to-date version, it is advisable to access the AIFA (Italian Medicines Agency) website. Disclaimer and useful information.
01.0 NAME OF THE MEDICINAL PRODUCT
MEDEOROS 35 MG TABLETS COATED WITH FILM
02.0 QUALITATIVE AND QUANTITATIVE COMPOSITION
Each tablet contains:
Active ingredient: 35 mg risedronate sodium (as 40.2 mg risedronate sodium hemipentahydrate)
Excipient with known effects: lactose.
For the full list of excipients, see section 6.1.
03.0 PHARMACEUTICAL FORM
Film-coated tablets.
Round, light orange tablets of 9 mm in diameter.
04.0 CLINICAL INFORMATION
04.1 Therapeutic indications
Treatment of postmenopausal osteoporosis to reduce the risk of vertebral fractures. Treatment of manifest postmenopausal osteoporosis to reduce the risk of hip fractures (see section 5.1).
Treatment of osteoporosis in men at high risk of fractures (see section 5.1).
04.2 Posology and method of administration
Dosage
The recommended dose for adults is one 35 mg tablet taken orally once a week. The tablet should be taken on the same day each week.
Pediatric population:
The use of risedronate sodium is not recommended in children below 18 years of age due to insufficient data on safety and efficacy (see also section 5.1).
Elderly patients:
No dosage adjustment is necessary as bioavailability, distribution and elimination in elderly subjects (> 60 years) were found to be similar to those in younger subjects. This was also demonstrated in very elderly patients, ie 75 years of age. and beyond in the postmenopausal population.
Impaired renal function:
No dosage adjustment is necessary in patients with mild to moderate renal impairment. The use of risedronate sodium is contraindicated in patients with severe renal impairment (creatinine clearance less than 30 ml / min) (see sections 4.3 and 5.2).
Method of administration
The absorption of risedronate sodium is affected by food and therefore, to ensure adequate absorption, patients should take risedronate sodium:
• before breakfast: at least 30 minutes before ingesting the first food, other medicinal products or drinks of the day (except for tap water).
Patients should be advised that if they forget to take a MEDEOROS 35 mg tablet they should take it on the day they remember. Patients should then resume taking one tablet per week on the day the tablet is usually taken. Two tablets should not be taken on the same day.
The tablet should be swallowed whole and not dissolved in the mouth or chewed. To facilitate the passage of the esophageal tablet, take risedronate sodium with a glass of tap water (≥120 ml), keeping the torso upright (standing or sitting). Once the tablet has been ingested, patients should avoid bedtime for 30 minutes (see section 4.4).
Calcium and vitamin D supplementation should be considered in case of inadequate dietary intake.
The optimal duration of bisphosphonate treatment for osteoporosis has not been established. The need for continued treatment should be reassessed in each individual patient periodically based on the potential benefits and risks, particularly after 5 or more years of use.
04.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
Hypocalcaemia (see section 4.4).
Pregnancy and breastfeeding.
Severe renal impairment (creatinine clearance
04.4 Special warnings and appropriate precautions for use
Food, drinks (except for tap water) and medicinal products containing polyvalent cations (such as calcium, magnesium, iron and aluminum) interfere with the absorption of bisphosphonates and should not be taken at the same time as risedronate sodium (see section 4.5 ).To achieve the desired efficacy, the administration instructions must be strictly adhered to (see section 4.2).
The efficacy of bisphosphonates in the treatment of postmenopausal osteoporosis is related to the presence of decreased bone mineral density and / or prevalence of fractures.
Older age or clinical risk factors for fractures alone do not justify initiating osteoporosis treatment with a bisphosphonate.
There is limited evidence to support the efficacy of bisphosphonates including risedronate sodium in very elderly women (over 80 years) (see section 5.1).
Bisphosphonates have been associated with esophagitis, gastritis, esophageal ulcers and gastroduodenal ulcers. Therefore, caution should be exercised:
• in patients with a history of esophageal disorders causing delayed esophageal transit or gastric emptying, such as narrowing or achalasia;
• in patients unable to keep their torso erect for at least 30 minutes from the time they take the tablet;
• if risedronate sodium is used in patients with current or recent problems with the upper gastrointestinal tract or the esophagus (including Barrett's esophagus).
Physicians should stress to patients the importance of heeding the administration instructions and be alert to any signs or symptoms that indicate a possible esophageal reaction. Patients should be advised that if they develop symptoms of esophageal irritation such as dysphagia, pain swallowing, retrosternal pain or the onset / aggravation of heartburn, should seek immediate medical attention.
Hypocalcaemia should be corrected prior to initiation of risedronate sodium therapy. It is also necessary to correct other disturbances of bone and mineral metabolism (e.g. parathyroid dysfunction, hypovitaminosis D) when initiating therapy with risedronate sodium.
Osteonecrosis of the jaw, usually associated with tooth extraction and / or local infection (including osteomyelitis), has been reported in cancer patients treated with regimens including bisphosphonates administered primarily intravenously. Many of these patients they were also treated with chemotherapy and corticosteroids. Osteonecrosis of the jaw has also been reported in patients with osteoporosis being treated with oral bisphosphonates.
Before starting treatment with bisphosphonates in patients with concomitant risk factors (such as cancer, chemotherapy, radiotherapy, corticosteroids, poor oral hygiene) the need for a dental examination with appropriate preventive dental procedures should be considered.
During treatment, these patients should, if possible, avoid invasive dental procedures. In patients who have developed osteonecrosis of the jaw and / or jaw during bisphosphonate therapy, dental surgery can exacerbate the condition. For patients requiring dental surgery, there are no data available to suggest that discontinuation of bisphosphonate treatment reduces the risk of osteonecrosis of the jaw and / or jaw.
The clinical judgment of the physician must guide the management program of each patient, based on the individual assessment of the risk / benefit ratio.
Osteonecrosis of the external auditory canal has been reported in conjunction with the use of bisphosphonates, predominantly in association with long-term therapy. Possible risk factors for osteonecrosis of the external auditory canal include the use of steroids and chemotherapy and / or local risk factors such as infection or trauma. Osteonecrosis of the external auditory canal should be considered in patients treated with bisphosphonates who have ear symptoms, including chronic ear infections.
Atypical fractures of the femur
Atypical subtrochanteric and shaft fractures of the femur have been reported, mainly in patients on long-term bisphosphonate therapy for osteoporosis. These short transverse or oblique fractures can occur anywhere in the femur from just below the lesser trochanter to above the supracondylar line. These fractures occur spontaneously or after minimal trauma and some patients experience thigh or groin pain, often associated with imaging findings and radiographic evidence of stress fractures, weeks or months before the onset of stress fractures. a complete femoral fracture. Fractures are often bilateral; therefore in bisphosphonate-treated patients who have sustained a femoral shaft fracture, the contralateral femur should be examined. Limited healing of these fractures has also been reported. In patients with suspected atypical femoral fracture, consideration should be given to discontinuing bisphosphonate therapy pending an assessment of the patient based on the individual benefit-risk ratio.
During treatment with bisphosphonates, patients should be advised to report any pain in the thigh, hip or groin and any patient who exhibits such symptoms should be evaluated for the presence of an incomplete fracture of the femur.
This drug contains lactose. Patients with rare hereditary problems of galactose intolerance, lactase deficiency or glucose-galactose malabsorption should not take this medicine.
04.5 Interactions with other medicinal products and other forms of interaction
No interaction studies with other treatments have been performed, however clinically relevant interactions with other medicinal products have not been observed in clinical trials. In phase III studies of risedronate sodium in the treatment of osteoporosis, 33% and 45% respectively of the patients took acetylsalicylic acid or other non-steroidal anti-inflammatory drugs (NSAIDs). In the phase III study with weekly dosing, 57% and 40% of postmenopausal patients received acetylsalicylic acid or other non-steroidal anti-inflammatory drugs, respectively. Among patients regularly treated with acetylsalicylic acid or NSAIDs (3 or more days per week), the incidence of upper gastrointestinal adverse events in patients treated with risedronate sodium was similar to that in the control group.
If deemed appropriate, risedronate sodium can be used concomitantly with estrogen replacement therapy (for women only).
The concomitant use of medicinal products containing polyvalent cations (eg calcium, magnesium, iron and aluminum) interferes with the absorption of risedronate sodium (see section 4.4).
Risedronate sodium is not systemically metabolised, does not induce cytochrome P-450 enzymes and is low in protein binding.
04.6 Pregnancy and breastfeeding
Pregnancy
There are insufficient data on the use of risedronate sodium in pregnant women. Studies in animals have shown reproductive toxicity (see section 5.3). The potential risk in women is unknown.
Feeding time
Animal studies indicate that a small amount of risedronate sodium passes into breast milk.
Risedronate sodium should not be administered in pregnant or breastfeeding women.
04.7 Effects on ability to drive and use machines
No effect on the ability to drive and use machines was observed.
04.8 Undesirable effects
Risedronate sodium has been studied in phase III clinical trials involving more than 15,000 patients.
Most of the undesirable effects seen in clinical trials were mild or moderate in severity and usually did not require discontinuation of therapy.
Undesirable effects that occurred during phase III clinical trials in women with postmenopausal osteoporosis treated for up to 36 months with risedronate sodium at a dose of 5 mg / day (n = 5020) or with placebo (n = 5048), and considered possibly or probably related to risedronate sodium, are listed using the following definition (the incidence versus placebo is indicated in parentheses):
Very common (≥1 / 10); common (≥1 / 100;
Nervous system disorders:
Common: headache (1.8% vs. 1.4%)
Eye disorders:
Uncommon: iritis *
Gastrointestinal disorders:
Common: constipation (5.0% vs. 4.8%), dyspepsia (4.5% vs. 4.1%), nausea (4.3% vs. 4.0%), abdominal pain (3.5 % vs. 3.3%), diarrhea (3.0% vs. 2.7%)
Uncommon: gastritis (0.9% vs. 0.7%), oesophagitis (0.9% vs. 0.9%), dysphagia (0.4% vs. 0.2%), duodenitis (0.2 % vs. 0.1%), oesophageal ulcer (0.2% vs. 0.2%)
Rare: glossitis (esophageal stricture (
Musculoskeletal and connective tissue disorders:
Common: musculoskeletal pain (2.1% vs. 1.9%).
Diagnostic tests:
Rare: liver function test abnormalities *
* No relevant incidence from Phase III clinical trials in osteoporosis; frequency is based on undesirable / laboratory / rechallenge data from previous clinical trials.
In a 1-year, double-blind, multicenter study comparing risedronate 5 mg daily (n = 480) and risedronate sodium 35 mg once weekly (n = 485) in postmenopausal women with osteoporosis, the overall tolerability and safety profiles were similar. The following additional undesirable effects considered by the investigator to be possibly or probably drug related (higher incidence in the risedronate 35 mg group than in the risedronate sodium 5 mg group) were reported: gastrointestinal disturbances (1.6% vs. 1.0%) and pain (1.2% vs. 0.8%).
In a 2-year multicenter study conducted in men with osteoporosis, the overall safety and tolerability profiles between the active therapy group and the placebo group were similar. The side effects matched those previously seen in women.
Laboratory parametersInitial mild, transient and asymptomatic decreases in serum calcium and phosphate have been observed in some patients.
The following additional undesirable effects have been reported from marketing: (frequency not known):
Eye disorders:
iritis, uveitis.
Musculoskeletal and connective tissue disorders:
osteonecrosis of the mandible and / or maxilla.
Skin and subcutaneous tissue disorders:
skin and hypersensitivity reactions, including angioedema, generalized rash, urticaria and bullous skin reactions and leukocytoclastic vasculitis, including some serious isolated cases of Stevens Johnson syndrome and toxic epidermal necrolysis.
Hair loss
Disorders of the immune system:
anaphylactic reactions.
Hepatobiliary disorders:
severe liver disease. In most of the reported cases, patients were also being treated with other products known to induce liver disease.
The following reactions have been reported during post-marketing experience (frequency rare): Atypical subtrochanteric and diaphyseal fractures of the femur (bisphosphonate class adverse reaction).
Very rare: osteonecrosis of the external auditory canal (adverse reaction for the bisphosphonate class).
Reporting of suspected adverse reactions
The reporting of suspected adverse reactions that occur after the authorization of the medicinal product is important, as it allows continuous monitoring of the benefit / risk ratio of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Italian Medicines Agency. at the address http://www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdose
No specific data are available on the treatment of cases of overdose with risedronate sodium.
In the event of an overdose, decreases in serum calcium can be expected. Some of these patients may also have signs and symptoms of hypocalcemia.
Milk or antacids containing magnesium, calcium or aluminum should be given to bind risedronate and reduce its absorption. In cases of overdose, gastric lavage may be considered to remove unabsorbed risedronate sodium.
05.0 PHARMACOLOGICAL PROPERTIES
05.1 Pharmacodynamic properties
Pharmaco-therapeutic category: Bisphosphonates, ATC code M05BA07.
Risedronate sodium is a pyridinyl bisphosphonate which attaches to the hydroxyapatite of the bone and inhibits bone resorption by osteoclasts. Bone turnover is reduced while osteoblastic activity and bone mineralization are maintained. In preclinical studies, risedronate sodium has shown a potent anti-osteoclastic and anti-resorption action resulting in a dose dependent increase in bone mass and biomechanical strength of bone. The activity of risedronate sodium was confirmed by measurements of biochemical indices of bone turnover during pharmacodynamic and clinical studies. In studies of postmenopausal women, decreases in biochemical indices of bone turnover were observed within the first month and reached the maximum level. within 3-6 months Decreases in these indices were similar with Risedronate 35 mg per week and Risedronate 5 mg / day after 12 months.
In a study in men with osteoporosis, decreases in biochemical indices of bone turnover were observed as early as 3 months and continued to be observed at 24 months.
Therapy and Prevention of Postmenopausal Osteoporosis :
Many risk factors including low bone mass, low bone mineral density, early menopause, smoking, and family history of osteoporosis are associated with postmenopausal osteoporosis. The clinical consequence of osteoporosis is the increased incidence of fractures. The risk of fractures increases with increasing risk factors.
Based on the effects on lumbar spine BMD, Risedronate 35 mg / week (n = 485) was shown to be equivalent to Risedronate 5 mg / day (n = 480) in a multi-center, double-blind study of duration of one year, in postmenopausal women with osteoporosis.
The daily dosing risedronate sodium clinical development program evaluated the effects of risedronate sodium on the risk of hip and vertebral fractures and included both early and late postmenopausal women with or without fractures. Doses of 2 were evaluated. , 5 and 5 mg per day and all groups, including controls, received calcium and vitamin D (if baseline levels were low). The absolute and relative risk of new vertebral and hip fractures was calculated by " use of an "analysis"time to first event'.
• Two placebo-controlled studies (n = 3,661) enrolled postmenopausal women less than 85 years of age with baseline vertebral fractures. Risedronate sodium 5 mg daily given for 3 years resulted in a reduction in the risk of new vertebral fractures compared with to the control group. In women with at least 2 vertebral fractures the relative risk reduction of new fractures was 49% (the incidence of new vertebral fractures with risedronate sodium was 18.1% and with placebo 29%) , in those with at least 1 fracture this reduction was 41%, (the incidence of new vertebral fractures with risedronate was 11.3% while with placebo it was 16.3%). The treatment effect was observed as early as the end of the first year of therapy. The benefits were also demonstrated in women with multiple fractures at baseline. Risedronate sodium 5 mg daily reduced annual loss compared to the control group.
• Two additional placebo-controlled studies enrolled postmenopausal women over 70 years of age with or without baseline vertebral fractures. Women aged 70-79 years with a skeletal femoral neck BMD T-score were enrolled for hip fracture or based on decreased mineral density of the femoral neck. Statistically, the efficacy of risedronate sodium versus placebo was only achieved when the two groups treated with 2.5 and 5 mg were combined. The following results are based on post-post analysis of patient subgroups only. chosen from clinical cases or on the current definition of osteoporosis:
• In a subgroup of patients with femoral neck BMD T-score ≤-2.5 SD (NHANES III) and with at least one baseline vertebral fracture, risedronate sodium administered for three years reduced the risk of hip fracture. hip fracture in 46% of cases compared to the control group (the incidence of hip fractures in the 2.5 and 5 mg risedronate sodium groups was 3.8%, 7.4% with placebo).
• The data suggest that more limited protection is evident in older patients (≥80 years). This may be a consequence of the increased importance of non-skeletal risk factors for hip fracture over the years. In these studies, the secondary endpoint analysis highlighted the decreased risk of new vertebral fractures in patients with decreased femoral neck BMD without vertebral fractures and in patients with decreased femoral neck BMD with or without vertebral fractures. .
• Risedronate sodium 5 mg daily given for 3 years increased bone mineral density (BMD) of the lumbar spine, femoral neck, trochanter and wrist compared to the control group and prevented bone loss in the distal third of the radio.
• A rapid reduction in the suppressive effects of risedronate sodium on bone turnover rate was observed in the "year following discontinuation of therapy after three years of treatment with risedronate sodium 5 mg daily."
• Bone biopsies performed on postmenopausal women treated with risedronate sodium 5 mg daily for 2-3 years confirmed the expected moderate decrease in bone turnover. Bone tissue during risedronate sodium treatment was found to have a normal lamellar structure and bone mineralization rate. These data, together with the decreased incidence of osteoporotic vertebral fractures in women with osteoporosis, seem to confirm the absence of harmful effects on bone quality.
Endoscopic measurements carried out on a number of patients, both on risedronate sodium and in the control group, suffering from various moderate to severe gastrointestinal disorders, did not reveal esophageal, gastric or duodenal ulcers related to the therapy, although cases of duodenitis were observed uncommonly in the risedronate sodium group.
Osteoporosis therapy in men
Risedronate sodium 35 mg once weekly was shown to be effective in men with osteoporosis (aged 36 to 84 years) in a 2-year, double-blind, placebo-controlled study in 284 patients (risedronate sodium 35 mg n = 191). All patients received calcium and vitamin D supplementation.
Increases in BMD were seen as early as 6 months after initiating treatment with risedronate sodium. Risedronate sodium 35 mg once weekly produced mean increases in BMD of the lumbar spine, femoral neck, trochanter and hip compared to placebo after 2 years of treatment. Anti-fracture efficacy was not demonstrated in this study.
The effect on bone (increased BMD and decreased biochemical markers of bone turnover) of risedronate sodium is similar in men and women.
Pediatric population
The safety and efficacy of risedronate sodium are being evaluated in an ongoing study in pediatric patients from 4 years to less than 16 years of age with osteogenesis imperfecta. Following completion of its randomized, double-blind, placebo-controlled phase, lasting one year, a statistically significant increase in lumbar spine BMD was demonstrated in the risedronate group versus the placebo group; however an increase in the number, of at least 1 new morphometric vertebral fracture (assessed radiographically), was found in the risedronate group compared to placebo. Overall, the results do not support the use of risedronate sodium in pediatric patients with osteogenesis imperfecta.
05.2 Pharmacokinetic properties
Absorption
Absorption of an oral dose is relatively rapid (Tmax ~ 1 hour) and is dose independent over the dose range studied (single dose study 2.5 to 30 mg; multiple dose studies 2.5 to 5 mg / day and up to 50 mg / week). The oral bioavailability of the tablet averages 0.63% and decreases when risedronate sodium is administered with food. Bioavailability was similar in men and women.
Distribution: the mean steady state volume of distribution in humans is 6.3 l / kg.
The fraction of the drug bound to plasma proteins is approximately 24%.
Biotransformation:
There is no evidence that risedronate sodium is metabolised systemically.
Elimination
Approximately half of the absorbed dose is eliminated in the urine within 24 hours, while 85% of an intravenous dose is eliminated in the urine after 28 days. Mean renal clearance is 105 ml / min and total clearance is 122 ml. / min: The difference is likely attributable to clearance due to absorption to bone. Renal clearance is not concentration dependent and there is a linear relationship between renal clearance and creatinine clearance. Unabsorbed risedronate sodium is eliminated unchanged via the faeces After oral administration the concentration-time curve shows three elimination phases with a terminal half-life of 480 hours.
Special populations
Elderly patients:
No dosage adjustment is necessary.
Patients treated with acetylsalicylic acid / NSAID:
Among patients treated regularly (three or more days a week) with acetylsalicylic acid or NSAIDs, the incidence of adverse events in the upper gastrointestinal tract in risedronate sodium was similar to that in the control group.
05.3 Preclinical safety data
Dose dependent hepatotoxic effects of risedronate sodium, mainly as an increase in enzymes, with histological changes in rats, were observed in toxicological studies in rats and dogs. The clinical relevance of these observations is unknown. Testicular toxicity appeared in rats and dogs at exposures considered in excess of the therapeutic exposure in humans. In rodents, dose-dependent irritation of the upper airways has often been noted. Similar effects have been reported with other bisphosphonates. Effects on the lower respiratory tract have been observed in long-term rodent studies, however the clinical relevance of these findings is unclear. In reproductive toxicity studies for exposures close to clinical exposures, changes in ossification at the sternal and / or cranial level were observed in fetuses of treated rats and hypocalcaemia and mortality in treated females who gave birth. There is no evidence of teratogenesis at delivery. dose of 3.2 mg / kg / day in rats and 10 mg / kg / day in rabbits, although data are only available on a limited number of rabbits. Maternal toxicity prevented the study of higher doses. on genotoxicity and carcinogenesis have not shown any particular risk for humans.
06.0 PHARMACEUTICAL INFORMATION
06.1 Excipients
Core: Microcrystalline cellulose, crospovidone, magnesium stearate, lactose monohydrate,
Coating: red iron oxide, yellow iron oxide, anhydrous colloidal silica, titanium dioxide, macrogol 400, macrogol 8000, hypromellose, hydroxypropyl cellulose.
06.2 Incompatibility
Not relevant.
06.3 Period of validity
3 years.
06.4 Special precautions for storage
This medicine does not require any special storage conditions.
06.5 Nature of the immediate packaging and contents of the package
Opaque PVC / PVDC / Aluminum blister in a cardboard box
Pack: 4 tablets
06.6 Instructions for use and handling
No special instructions.
07.0 MARKETING AUTHORIZATION HOLDER
FENIX PHARMA SOC. COOPERATIVE
Via Ercolano Salvi n. 18
00143 Rome
Italy
08.0 MARKETING AUTHORIZATION NUMBER
AIC n. 040044012 "MEDEOROS 35 mg film-coated tablets - 4 tablets"
09.0 DATE OF FIRST AUTHORIZATION OR RENEWAL OF THE AUTHORIZATION
Date of first authorization: 20 June 2011
Latest renewal date:
10.0 DATE OF REVISION OF THE TEXT
February 2016