Active ingredients: Zoledronic acid
Aclasta 5 mg solution for infusion
Why is Aclasta used? What is it for?
Aclasta contains the active substance zoledronic acid. It belongs to a group of medicines called bisphosphonates and is used to treat postmenopausal women and adult men with osteoporosis or osteoporosis caused by treatment with corticosteroids used to treat inflammation, and Paget's disease of bone in adults.
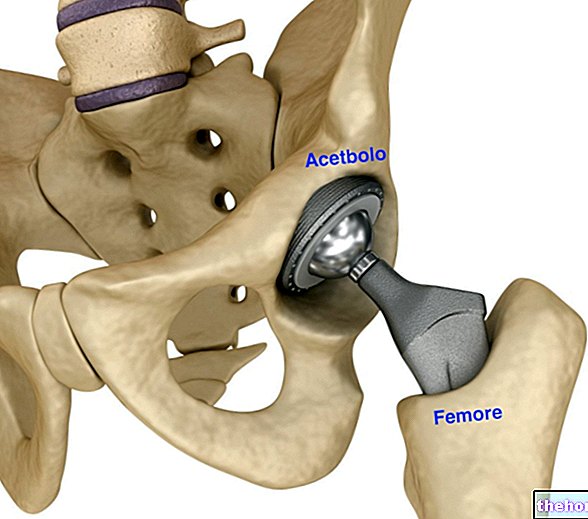
Osteoporosis
Osteoporosis is a disease that causes thinning and weakening of the bones and is common in women after menopause but can also occur in men. At the time of menopause, the ovaries stop producing the female hormone estrogen, which contributes to preserve the state of health of the bones. Bone loss occurs following menopause, bones become weaker and break more easily. Osteoporosis can also occur in men and women due to long-term steroid use which can affect bone strength. Many patients with osteoporosis have no symptoms, but are still at risk of bone fracture as osteoporosis has made their bones more fragile. The reduction in the levels of circulating sex hormones, mainly estrogen converted from androgens, also plays a role in the more gradual bone loss seen in men. In both women and men, Aclasta strengthens the bones and makes the risk of fracture less likely. Aclasta is also used in patients who have recently had a hip fracture due to minor trauma such as a fall and are therefore at risk for bone fractures.
Paget's disease of the bone
It is normal for aged bone to be removed and replaced by new bone. This process is called bone remodeling. In Paget's disease, bone remodeling is too rapid and new bone forms in a disorderly fashion, which makes it weaker than normal. If the disease is not treated, the bones can deform and become painful, and they can break. Aclasta works to bring the bone remodeling process back to normal, ensuring normal bone formation, thus restoring bone strength.
Contraindications When Aclasta should not be used
Follow carefully all instructions given to you by your doctor, pharmacist or nurse before you are given Aclasta.
Aclasta must not be given to you:
- if you are allergic to zoledronic acid, other bisphosphonates or any of the other ingredients of this medicine
- if you have hypocalcaemia (i.e. if your blood calcium levels are too low)
- if you have severe kidney problems
- if you are pregnant.
- if you are breastfeeding.
Precautions for use What you need to know before taking Aclasta
Talk to your doctor before you are given Aclasta:
- if you are being treated with any medicine containing zoledronic acid, which is also the active substance in Aclasta (zoledronic acid is used in adult patients with certain cancers to prevent bone complications or to reduce the amount of calcium)
- if you have kidney problems, or have ever had any
- if you cannot take a daily calcium supplement
- if you have had part or all of the parathyroid glands in your neck removed by surgery.
- if you have had sections of your intestine removed.
An undesirable effect called osteonecrosis of the jaw (bone damage of the jaw) has been reported in post-marketing experience in patients treated with Aclasta (zoledronic acid) for the treatment of "" osteoporosis. Osteonecrosis of the jaw. it can also occur after stopping treatment.
It is important to try to prevent the onset of osteonecrosis of the jaw as it is a painful condition that can be difficult to treat. To reduce the risk of developing osteonecrosis of the jaw there are some precautions you must take.
Before receiving treatment with Aclasta, talk to your doctor, pharmacist or nurse if:
- have any problems with your mouth or teeth such as poor dental health, gum disease, or have planned a "tooth extraction
- do not receive routine dental care or have not had a dental check-up in a long time
- you are a smoker (as this may increase the risk of dental problems)
- have previously been treated with a bisphosphonate (used to treat or prevent bone disorders);
- are taking medicines called corticosteroids (such as prednisolone or dexamethasone)
- has cancer.
Your doctor may ask you to have a dental examination before starting treatment with Aclasta.
During treatment with Aclasta, you must maintain good oral hygiene (which includes regular teeth brushing) and have routine dental check-ups. If you wear dentures, you need to make sure they are properly secured. If you are having dental treatment or are due to undergo dental surgery (e.g. tooth extractions), please tell your doctor and tell your dentist that you are being treated with Aclasta. Tell your doctor and dentist immediately if you experience any problems with your mouth or teeth such as loosening, pain, swelling or non-healing sores or discharge, as these may be signs of osteonecrosis of the jaw.
Monitoring test
Your doctor must take a blood sample to check your kidney function (creatinine levels) before each infusion of Aclasta. It is important that you drink at least two glasses of liquids (eg water) within a few hours before your Aclasta treatment, according to the caregiver's instructions.
Children and adolescents
Aclasta is not recommended under 18 years of age. The use of Aclasta in children and adolescents has not been studied
Interactions Which drugs or foods may change the effect of Aclasta
Tell your doctor, pharmacist or nurse if you are taking, have recently taken, might take any other medicines.
It is important for your doctor to know all the medicines you are taking, especially if you are already taking other medicines that are potentially harmful to the kidneys (eg aminoglycosides) or diuretics ('medicines to urinate') which can cause dehydration.
Warnings It is important to know that:
Pregnancy and breastfeeding
You must not be given Aclasta if you are pregnant or breast-feeding, think you may be pregnant or are planning to become pregnant.
Ask your doctor, pharmacist or nurse for advice before taking this medicine.
Driving and using machines
If you feel dizzy while taking Aclasta, do not drive or use machines until you feel better.
Aclasta contains sodium
This medicinal product contains less than 1 mmol sodium (23 mg) per 100 ml bottle of Aclasta, thus essentially "sodium-free".
Dose, Method and Time of Administration How to use Aclasta: Posology
Carefully follow all instructions given to you by your doctor or nurse. If in doubt, consult your doctor or nurse.
Osteoporosis
The usual dose is 5 mg, given by your doctor or nurse as a single infusion into your vein per year. The infusion will last at least 15 minutes.
In the case of a recent hip fracture, it is recommended that Aclasta be administered two or more weeks after hip fracture surgery.
It is important to take calcium and vitamin D supplements (e.g. tablets) according to your doctor's instructions.
For osteoporosis, Aclasta works for one year. Your doctor will let you know when to come back for your next dose.
Paget's disease
For the treatment of Paget's disease, Aclasta should only be prescribed by doctors experienced in the treatment of Paget's disease of the bone.
The usual dose is 5 mg, given by your doctor or nurse in an initial infusion into a vein. The infusion will last at least 15 minutes. Aclasta can work for more than a year and your doctor will let you know if you need another treatment.
Your doctor may advise you to take calcium and vitamin D supplements (e.g. tablets) for at least the first ten days after taking Aclasta. It is important that you follow this advice carefully so that your blood calcium level is not too low in the period following the infusion. Your doctor will inform you about the possible symptoms associated with hypocalcemia.
Aclasta with food and drink
Make sure you drink enough fluids (at least one or two glasses) before and after Aclasta treatment as directed by your doctor. This will help prevent dehydration. You can eat normally on the day of your Aclasta treatment. This is especially important in patients taking diuretics (urinating pills) and in elderly patients (65 years of age or older).
If you forget a dose of Aclasta
Contact your doctor or hospital as soon as possible in order to make a new appointment.
Before stopping treatment with Aclasta
If you are considering stopping Aclasta treatment, please come to your next appointment and discuss this with your doctor. Your doctor can advise you and decide how long to continue the treatment.
If you have any further questions on the use of this medicine, ask your doctor, pharmacist or nurse.
Side Effects What are the side effects of Aclasta
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Side effects related to the first infusion are very common (occurring in more than 30% of patients) but are less common following subsequent infusions. Most side effects such as fever and chills, pain in muscles or joints and headache occur in the first three days after taking Aclasta. Symptoms are usually mild to moderate and disappear within three days. Your doctor may recommend a mild pain reliever such as ibuprofen or acetaminophen to reduce these side effects. The chance of having side effects decreases with subsequent doses of Aclasta.
Some side effects can be serious
Common (may affect up to 1 in 10 people)
Irregular heart rhythm (atrial fibrillation) has been observed in patients being treated with Aclasta for postmenopausal osteoporosis. It is currently unclear whether Aclasta is the cause of this irregular heart rhythm but you should tell your doctor if after having administered Aclasta exhibits such symptoms.
Uncommon (may affect up to 1 in 100 people)
Swelling, redness, pain and itching of the eyes or sensitivity of the eyes to light.
Very rare (may affect up to 1 in 10,000 people)
Talk to your doctor if you have ear pain, ear discharge and / or ear infection. These episodes could be signs of bone damage in your ear. Not known (frequency cannot be estimated from the available data) Pain in the mouth and / or jaw, swelling or sores in the mouth or jaw that do not heal, discharge, numbness or a feeling of heaviness in the jaw or loosening of a tooth ; these could be signs of severe bone degeneration of the jaw (osteonecrosis). Tell your doctor and dentist immediately if you experience such symptoms while being treated with Aclasta or after stopping treatment.
Kidney disorders (e.g. decreased amount of urine) may occur. Your doctor will need to draw blood to check your kidney function before each infusion of Aclasta. It is important that you drink at least one or two glasses of liquids (eg water) within a few hours before your Aclasta treatment, as directed by your healthcare provider.
If you get any of these side effects, tell your doctor immediately.
Aclasta can cause other side effects as well
Very common (may affect more than 1 in 10 people)
Fever
Common (may affect up to 1 in 10 people)
Headache, dizziness, malaise, vomiting, diarrhea, body aches, bone and / or joint pain, pain in the back, arms or legs, flu-like symptoms (eg tiredness, chills, joint and muscle pain), chills , feeling tired and lacking of interest, weakness, pain, feeling unwell, swelling and / or pain at the infusion site.
In patients with Paget's disease, symptoms of low blood calcium, such as muscle spasms, or numbness, or tingling especially in the area around the mouth have been reported.
Uncommon (may affect up to 1 in 100 people)
Influenza, upper respiratory tract infections, decreased red blood cell count, loss of appetite, insomnia, sleepiness which may include decreased alertness and consciousness, tingling or numbness, extreme tiredness, tremor, temporary loss of consciousness, eye infection or irritation or inflammation with pain and redness, feeling dizzy, increased blood pressure, flushing, cough, shortness of breath, stomach pain, abdominal pain, constipation, dry mouth, heartburn, rash, excessive sweating , itching, skin redness, neck pain, muscle, bone and / or joint stiffness, joint swelling, muscle spasms, shoulder pain, chest and chest muscle pain, joint inflammation, muscle weakness, abnormal results kidney tests, frequent abnormal urge to urinate, swelling of the hands, ankles or feet, thirst, toothache, high taste ration.
Rare (may affect up to 1 in 1000 people)
Rarely, particularly in patients on long-term treatment for osteoporosis, an unusual fracture of the femur may occur. Contact your doctor if you experience pain, weakness or discomfort in the thigh, hip or groin as it could be a " early indication of a possible fracture of the femur.
Not known (frequency cannot be estimated from the available data)
severe allergic reactions including dizziness and difficulty in breathing, swelling mainly of the face and throat, decreased blood pressure, dehydration secondary to post-infusion symptoms such as fever, vomiting and diarrhea.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly via the national reporting system listed in Appendix V. side effects you can help provide more information on the safety of this medicine.
Expiry and Retention
Your doctor, pharmacist or nurse are advised on how to properly store Aclasta.
- Keep this medicine out of the sight and reach of children.
- Do not use this medicine after the expiry date which is stated on the carton and bottle after EXP.
- The unopened bottle does not require any special storage conditions.
- After opening the bottle, the product should be used immediately in order to avoid microbial contamination. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user and would normally not be longer than 24 hours at 2 ° C - 8 ° C. Wait for the solution taken from the refrigerator to reach room temperature before use.
Other Information
What Aclasta contains
The active substance is zoledronic acid. Each 100 ml bottle of solution contains 5 mg of zoledronic acid (as monohydrate). One ml of solution contains 0.05 mg of zoledronic acid (as monohydrate).
The other ingredients are mannitol, sodium citrate and water for injections.
What Aclasta looks like and contents of the pack
Aclasta is a clear and colorless solution. It comes in 100 ml plastic bottles as a ready-to-infuse solution. It is supplied in cartons containing one bottle for single pack or in multi-dose cartons comprising five packs, one bottle each. Not all pack sizes may be marketed.
Source Package Leaflet: AIFA (Italian Medicines Agency). Content published in January 2016. The information present may not be up-to-date.
To have access to the most up-to-date version, it is advisable to access the AIFA (Italian Medicines Agency) website. Disclaimer and useful information.
01.0 NAME OF THE MEDICINAL PRODUCT
ACLASTA 5 MG SOLUTION FOR INFUSION
02.0 QUALITATIVE AND QUANTITATIVE COMPOSITION
Each bottle with 100 ml of solution contains 5 mg of zoledronic acid (as monohydrate).
Each ml of the solution contains 0.05 mg of zoledronic acid (as monohydrate).
For the full list of excipients, see section 6.1.
03.0 PHARMACEUTICAL FORM
Solution for infusion
Clear and colorless solution.
04.0 CLINICAL INFORMATION
04.1 Therapeutic indications
Treatment of osteoporosis
• in postmenopausal women
• in adult men
at increased risk of fractures, including those with a recent mild trauma hip fracture.
Treatment of osteoporosis associated with long-term systemic glucocorticoid therapy
• in postmenopausal women
• in adult men
at increased risk of fracture.
Treatment of Paget's disease of bone in adults.
04.2 Posology and method of administration
Dosage
Patients must be adequately hydrated prior to administration of Aclasta. This is particularly important for the elderly (≥65 years) and for patients on diuretic therapy.
It is recommended to combine the administration of Aclasta with an adequate supplement of calcium and vitamin D.
Osteoporosis
For the treatment of postmenopausal osteoporosis, osteoporosis in humans and for the treatment of osteoporosis associated with long-term systemic glucocorticoid therapy, the recommended dose is a single intravenous infusion of Aclasta 5 mg administered once daily. The optimal duration of bisphosphonate treatment for osteoporosis has not been established. The need for continued treatment should be reassessed in each individual patient periodically based on the potential benefits and risks of Aclasta, particularly after 5 or more years of use. In patients with a recent mild trauma hip fracture, it is recommended administration by infusion of Aclasta at least two weeks after hip fracture healing (see section 5.1). In patients with a recent mild trauma hip fracture, a loading dose of 50,000 to 125,000 IU is recommended. vitamin D, administered orally or intramuscularly, prior to the first infusion of Aclasta.
Paget's disease
For the treatment of Paget's disease, Aclasta should only be prescribed by doctors experienced in the treatment of Paget's disease of the bone. The recommended dose is a single intravenous infusion of Aclasta 5 mg. Patients with Paget's disease are strongly advised to ensure adequate calcium supplement corresponding to at least 500 mg of elemental calcium twice daily for at least 10 days following Aclasta administration (see section 4.4).
Retreatment of Paget's disease: In Paget's disease, a prolonged remission period was observed in responding patients following initial treatment with Aclasta. Retreatment in relapsing patients consists of an "additional intravenous infusion of Aclasta 5 mg after an interval of one year or more from initial treatment. Limited data are available on retreatment of Paget's disease (see section 5.1).
Special populations
Patients with renal impairment
Aclasta is contraindicated in patients with creatinine clearance
No dose adjustment is required in patients with creatinine clearance ≥35 ml / min.
Patients with hepatic impairment
No dose adjustment is required (see section 5.2).
Elderly (≥65 years old)
Since bioavailability, distribution and elimination were similar in elderly and younger subjects, no dose adjustment is necessary.
Pediatric population
The safety and efficacy of Aclasta in children and adolescents below 18 years have not been established. There are no data available.
Method of administration
Intravenous use.
Aclasta is administered through an infusion line with a ventilating membrane and is administered slowly at a constant infusion rate. The infusion time should not be less than 15 minutes. For information on how Aclasta is infused, see section 6.6.
Patients treated with Aclasta should be given the package leaflet and the patient reminder card.
04.3 Contraindications
- Hypersensitivity to the active substance, to any bisphosphonate or to any of the excipients listed in section 6.1.
- Patients with hypocalcaemia (see section 4.4).
- Severe renal impairment with creatinine clearance
- Pregnancy and lactation (see section 4.6).
04.4 Special warnings and appropriate precautions for use
Kidney function
The use of Aclasta in patients with severe renal impairment (creatinine clearance renal insufficiency in this population.
Renal impairment has been observed following administration of Aclasta (see section 4.8) particularly in patients with pre-existing renal dysfunction or with other risk factors including advanced age, concomitant use of nephrotoxic medicinal products, concomitant diuretic therapy (see section 4.5). , or dehydration following the administration of Aclasta. Renal impairment has been observed in patients after single administration. Renal failure involving the need for dialysis or with a fatal outcome has occurred rarely in patients with underlying renal impairment or with any of the risk factors described above. To minimize the risk of renal adverse reactions the following precautions should be considered:
• Before each infusion of Aclasta, creatinine clearance from body weight should be calculated using the Cockcroft-Gault formula.
• The transient increase in serum creatinine may be more pronounced in patients with underlying renal impairment.
• Periodic monitoring of serum creatinine should be considered in patients at risk.
• Aclasta should be used with caution when administered concomitantly with other medicinal products that may impact renal function (see section 4.5).
• Patients, particularly elderly patients and those taking diuretics, should be adequately hydrated prior to administration of Aclasta.
• A single infusion of Aclasta should not exceed 5 mg and the duration of the infusion should be at least 15 minutes (see section 4.2).
Hypocalcemia
Pre-existing hypocalcaemia should be treated with adequate administration of calcium and vitamin D before initiating therapy with Aclasta (see section 4.3). Other alterations in mineral metabolism must also be adequately treated, (e.g. reduced parathyroid reserve, intestinal calcium malabsorption). For these patients, physicians should evaluate the possibility of clinical monitoring.
High bone turnover is a feature of Paget's disease of the bone. Due to the rapid onset of the effect of zoledronic acid on bone turnover, transient, sometimes symptomatic, hypocalcaemia may develop, reaching maximum levels usually within 10 days following the Aclasta infusion (see section 4.8).
It is recommended to combine the administration of Aclasta with an adequate calcium and vitamin D supplement. In addition, patients suffering from Paget's disease are strongly advised to ensure an adequate calcium supplement corresponding to at least 500 mg of calcium twice a day at least in 10 days following Aclasta administration (see section 4.2). Patients should be informed of the possible symptoms caused by hypocalcaemia and should be monitored appropriately from a clinical point of view during the risk period. In patients with Paget's disease it is recommended that serum calcium be measured prior to the Aclasta infusion. Severe and occasionally disabling bone, joint and / or muscle pain has been reported infrequently in patients receiving bisphosphonates, including zoledronic acid (see section 4.8).
Osteonecrosis of the mandible / maxilla
Osteonecrosis of the jaw has been reported in post-marketing experience in patients treated with Aclasta (zoledronic acid) for osteoporosis (see section 4.8). The initiation of treatment or a new course of treatment should be postponed in patients with unhealed open lesions of the soft tissues of the oral cavity. A dental examination with concomitant risk factors is recommended before starting treatment with Aclasta in patients with concomitant risk factors. appropriate preventive dental procedures and an individual benefit-risk assessment When evaluating the risk for a patient of developing osteonecrosis of the jaw, the following should be considered:
- The potency to inhibit bone resorption of the drug (higher risk for very potent molecules), route of administration (higher risk for parenteral administration) and cumulative dose.
- Cancer, co-morbidities (eg: anemia, coaugulopathies, infection), smoking.
- Concomitant therapies: corticosteroids, chemotherapy, angiogenesis inhibitors, head and neck radiotherapy.
- Poor oral hygiene, periodontal disease, poorly fixed dentures, history of dental disease, invasive dental procedures, eg: tooth extractions.
All patients should be encouraged to maintain good oral hygiene, undergo routine dental check-ups, and immediately report any oral symptoms such as tooth mobility, pain, swelling or non-healing of sores, or discharge during treatment with zoledronic acid. In the course of treatment, invasive dental procedures should be performed with caution and avoided in close proximity to zoledronic acid treatment.
The management program for patients who develop osteonecrosis of the jaw should be established in close collaboration between the treating physician and a dentist or oral surgeon competent in osteonecrosis of the jaw. Temporary interruption of zoledronic acid treatment should be considered until the condition resolves and concomitant risk factors are mitigated where possible.
Osteonecrosis of the external auditory canal
Osteonecrosis of the external auditory canal has been reported in conjunction with the use of bisphosphonates, predominantly in association with long-term therapy. Possible risk factors for osteonecrosis of the external auditory canal include the use of steroids and chemotherapy and / or local risk factors such as infection or trauma. Osteonecrosis of the external auditory canal should be considered in patients treated with bisphosphonates who have ear symptoms, including chronic ear infections.
Atypical fractures of the femur
Atypical subtrochanteric and diaphyseal fractures of the femur have been reported, mainly in patients on long-term bisphosphonate therapy for osteoporosis. These short transverse or oblique fractures can occur anywhere in the femur from just below the lesser trochanter to above the supracondylar line. These fractures occur spontaneously or after minimal trauma and some patients experience thigh or groin pain, often associated with imaging evidence of stress fractures, weeks or months before a hip fracture occurs. complete. Fractures are often bilateral; therefore in bisphosphonate-treated patients who have sustained a femoral shaft fracture, the contralateral femur should be examined. Limited healing of these fractures has also been reported. In patients with suspected atypical femoral fracture, discontinuation of bisphosphonate therapy should be considered pending an assessment of the patient based on individual benefit-risk.
During treatment with bisphosphonates, patients should be advised to report any pain in the thigh, hip or groin and any patient who exhibits such symptoms should be evaluated for the presence of an incomplete fracture of the femur.
General
The incidence of post-infusion symptoms occurring within the first three days following Aclasta administration can be reduced by administering paracetamol or ibuprofen immediately following Aclasta administration.
Other products are available containing zoledronic acid as an active substance for oncological indications.Patients treated with Aclasta should not be treated concomitantly with these products or any other bisphosphonates, as the combined effects of these substances are unknown. This medicinal product contains less than 1 mmol sodium (23 mg) per 100 ml bottle of Aclasta, thus essentially "sodium-free".
04.5 Interactions with other medicinal products and other forms of interaction
No interaction studies with other medicinal products have been performed. Zoledronic acid is not systemically metabolised and does not affect in vitro the activity of human cytochrome P450 enzymes (see section 5.2). Zoledronic acid does not bind extensively to plasma proteins (approximately 43-55% of the drug is bound) and therefore interactions resulting from the displacement of medicinal products with high protein binding.
Zoledronic acid is eliminated by renal excretion. Use caution if zoledronic acid is administered in combination with medicinal products which may have a significant impact on renal function (eg aminoglycosides or diuretics which may cause dehydration) (see section 4.4).
In patients with renal impairment, systemic exposure to concomitantly administered medicinal products excreted primarily via the kidney may be increased.
04.6 Pregnancy and breastfeeding
Women of childbearing potential
Aclasta is not recommended in women of childbearing age.
Pregnancy
Aclasta is contraindicated during pregnancy (see section 4.3). There are no adequate data from the use of zoledronic acid in pregnant women. Animal studies with zoledronic acid have shown reproductive toxicity including malformations (see section 5.3). The potential risk for humans is unknown.
Feeding time
Aclasta is contraindicated during lactation (see section 4.3). It is not known whether zoledronic acid is excreted in human milk.
Fertility
Zoledronic acid was evaluated in rats for potential adverse effects on the fertility of the parents and the F1 generation. This resulted in pronounced pharmacological effects considered to be related to the inhibition of skeletal calcium mobilization by the compound, resulting in hypocalcemia during peripartum, a bisphosphonate class effect, dystocia and early termination of the study. These results therefore do not allow to determine a definitive effect of Aclasta on fertility in humans.
04.7 Effects on ability to drive and use machines
Adverse reactions, such as dizziness, may affect the ability to drive or use machines.
04.8 Undesirable effects
Summary of the safety profile
The overall percentage of patients experiencing adverse reactions was 44.7%, 16.7% and 10.2% after the first, second and third infusions, respectively. The incidence of individual adverse reactions following the first infusion was: pyrexia (17.1%), myalgia (7.8%), flu-like illness (6.7%), arthralgia (4.8%) and headache (5.1%) The incidence of these reactions decreased markedly with successive annual doses of Aclasta. Most of these reactions occurred in the first three days following Aclasta administration. Most of these reactions were mild to moderate and resolved within three days of the occurrence of the event. In a smaller study where prophylaxis of adverse reactions was performed as described below, the percentage of patients who experienced adverse reactions was lower (19.5%, 10.4%, 10.7% after the first, second and third infusions, respectively).
Table of adverse reactions
Adverse reactions in Table 1 are listed by MedDRA system organ class and frequency category. Frequency categories are defined using the following convention: very common (≥1 / 10); common (≥1 / 100,
Table 1
# Observed in patients taking concomitant glucocorticoids.
* Common only in Paget's disease.
** Based on post-marketing reports. Frequency cannot be estimated from the available data.
† Identified during post-marketing experience.
Description of selected adverse reactions
Atrial fibrillation
In the HORIZON - Pivotal Fracture Trial [PFT] (see section 5.1), the overall incidence of atrial fibrillation was 2.5% (96 out of 3,862) and 1.9% (75 out of 3,852) in treated patients, respectively. with Aclasta and placebo The rate of serious adverse events of atrial fibrillation increased in patients taking Aclasta (1.3%) (51 out of 3,862) compared to patients receiving placebo (0.6%) (22 out of 3,852). The mechanism behind the increased incidence of atrial fibrillation is unknown. In the osteoporosis studies (PFT, HORIZON - Recurrent Fracture Trial [RFT]) the combined incidence of atrial fibrillation was comparable between Aclasta (2.6%) and placebo (2.1%). For serious adverse events of atrial fibrillation the combined incidence was 1.3% for Aclasta and 0.8% for placebo.
Class Effects:
Renal impairment
Zoledronic acid has been associated with renal impairment evidenced by deterioration of renal function (ie increased serum creatinine) and in rare cases by acute renal failure. Following administration of zoledronic acid, mainly in patients with pre-existing renal dysfunction or with additional risk factors (e.g. older age, cancer patients undergoing chemotherapy, concomitant use of nephrotoxic medicinal products, concomitant diuretic therapy, severe dehydration) renal impairment was observed. In the majority of cases these patients were being treated with a dose of 4 mg each 3-4 weeks, but the alteration was also detected after a single administration.
In clinical trials in osteoporosis, changes in creatinine clearance (measured annually prior to dosing) and the incidence of renal insufficiency and impairment were comparable in both the Aclasta and placebo treatment groups over three years. There was a transient increase in serum creatinine observed in the first 10 days in 1.8% of patients treated with Aclasta compared with 0.8% of patients treated with placebo.
Hypocalcemia
In clinical trials of osteoporosis, approximately 0.2% of patients showed a considerable decrease in serum calcium levels (less than 1.87 mmol / l) following administration of Aclasta. No symptomatic cases of hypocalcaemia were observed. .
In Paget's disease studies, symptomatic hypocalcaemia was observed in approximately 1% of patients, with receding in all cases.
Based on laboratory values, asymptomatic transient calcium levels below the normal reference range (less than 2.10 mmol / L) occurred in 2.3% of patients treated with Aclasta in a large clinical trial compared 21% of patients treated with Aclasta in Paget's disease studies. The frequency of hypocalcaemia was much lower following subsequent infusions.
Adequate vitamin D and calcium supplementation was administered to all patients enrolled in the postmenopausal osteoporosis study, the clinical fracture prevention study after hip fracture and the Paget's disease studies (see also section 4.2). In the clinical fracture prevention study after a recent hip fracture, vitamin D levels were not routinely measured but the majority of patients received a loading dose of vitamin D prior to administration of Aclasta (see paragraph 4.2).
Local reactions
In a large clinical study local reactions at the infusion site (0.7%) such as redness, swelling and / or pain were reported after administration of zoledronic acid.
Osteonecrosis of the mandible / maxilla
Cases of osteonecrosis (of the jaw) have been reported mainly in cancer patients treated with products that inhibit bone resorption, including zoledronic acid (see section 4.4). In a large clinical study in 7,736 patients it is Osteonecrosis of the jaw has been reported in one patient treated with Aclasta and in one treated with placebo. Cases of osteonecrosis of the jaw have been reported in the post-marketing experience of Aclasta.
Reporting of suspected adverse reactions
Reporting of suspected adverse reactions occurring after authorization of the medicinal product is important as it allows continuous monitoring of the benefit / risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system.
04.9 Overdose
Clinical experience with acute overdose is limited. Patients who have been treated with doses higher than those recommended should be monitored with particular care. an oral calcium and / or intravenous calcium gluconate supplement.
05.0 PHARMACOLOGICAL PROPERTIES
05.1 Pharmacodynamic properties
Pharmacotherapeutic group: Drugs for the treatment of bone diseases, bisphosphonates, ATC code: M05BA08
Mechanism of action
Zoledronic acid belongs to the class of nitrogen-containing bisphosphonates and acts primarily on bone tissue. It is an inhibitor of the osteoclast-mediated bone resorption process.
Pharmacodynamic effects
The selective action of bisphosphonates on bone is due to their high affinity for mineralized bone. The main molecular target of zoledronic acid is the enzyme farnesyl pyrophosphate synthetase in osteoclast. The long duration of action of zoledronic acid is attributable to its high binding affinity for the active site of farnesyl pyrophosphate (FPP) synthetase and its strong affinity towards mineralized bone.
Aclasta treatment rapidly reduced the rate of bone turnover from high postmenopausal levels with the nadir of resorption markers observed at day 7 and markers of formation at week 12. Thereafter, bone markers stabilized in the premenopausal ranges. There was no progressive reduction in bone turnover markers with repeated annual dosing.
Clinical efficacy in the treatment of postmenopausal osteoporosis (PFT)
The efficacy and safety of Aclasta 5 mg once yearly for 3 consecutive years has been demonstrated in postmenopausal women (7,736 women aged 65-89 years) with: Bone Mineral Density (BMD) T-score femoral neck ≤ -1.5 and at least two pre-existing mild or one moderate vertebral fractures; or Femoral neck BMD T-score ≤ -2.5 with or without evidence of pre-existing vertebral fractures. 85% of patients were on first bisphosphonate treatment. Women evaluated for vertebral fracture incidence did not receive concomitant osteoporosis therapy, which was given to women evaluated for hip fractures and all fractures. clinics. Concomitant therapy for osteoporosis included: calcitonin, raloxifene, tamoxifen, hormone replacement therapy, tibolone; but excluded other bisphosphonates. All women received 1,000 to 1,500 mg of elemental calcium and 400 to 1,200 IU supplement daily. of vitamin D.
Effect on morphometric vertebral fractures
Aclasta significantly reduced the incidence of one or more new vertebral fractures over three years and as early as the first year survey (see Table 2).
Table 2 Summary of efficacy in vertebral fractures at 12, 24 and 36 months
Patients aged 75 years or older treated with Aclasta showed a 60% reduction in the risk of vertebral fractures compared to patients treated with placebo (p
Effect on hip fractures
Aclasta demonstrated a consistent effect over 3 years resulting in a 41% reduction in the risk of hip fractures (95% CI, 17% to 58%). The hip fracture episode rate was 1.44% in the Aclasta group, compared with 2.49% in the placebo group. The risk reduction was 51% in patients on first bisphosphonate treatment and 42% in patients granted concomitant osteoporosis therapy.
Effect on all clinical fractures
All clinical fractures were examined based on radiographic and / or clinical evidence. A summary of the results is presented in Table 3.
Table 3 Comparison of treatments in the incidence of major clinical fracture variables over 3 years
Effect on bone mineral density (BMD)
Aclasta significantly increased lumbar spine, hip and distal radius BMD relative to placebo treatment across all timepoints (6, 12, 24 and 36 months). Treatment with Aclasta showed a 6.7% increase in lumbar spine BMD, 6.0% total hip, 5.1% femoral neck and 3.2% distal radius compared to placebo. in the 3 years of treatment.
Bone histology
In 152 postmenopausal patients with osteoporosis treated with Aclasta (N = 82) and placebo (N = 70), bone biopsies were obtained from the iliac crest 1 year after the third annual dose. Histomorphometric analysis showed a 63% reduction in bone turnover. Osteomalacia, cystic fibrosis and formation of woven bone. With the exception of one case, the tetracycline marker was found in all 82 biopsies performed on patients treated with Aclasta. Microcomputerized tomography (µCT) demonstrated an increase in trabecular bone volume and maintenance of the architecture of the trabecular bone. "Trabecular bone in patients treated with Aclasta compared to the placebo group.
Bone turnover marker
Evaluations of bone-specific alkaline phosphatase (BALP), serum N-terminal collagen type I propeptide (P1NP), and serum beta-C telopeptides (b-CTx) in subgroups of 517 to 1,246 patients at periodic intervals throughout the study. . Treatment with a 5 mg annual dose of Aclasta significantly reduced BALP by 30% from baseline at 12 months, which was maintained at 28% below baseline at 36 months. P1NP significantly decreased 61% below the 12-month baseline level and remained 52% below the 36-month baseline level. B-CTx was significantly reduced by 61% from baseline at 12 months and remained 55% below baseline levels at 36 months. Throughout the time period observed, bone turnover markers remained within the pre-menopausal range at the end of each year. Repeat dosing did not result in further reductions in bone turnover markers.
Effect on height
In the three-year osteoporosis study, standing height was measured annually with the aid of a stadiometer. The Aclasta-treated group experienced approximately 2.5 mm less stature reduction than the placebo group (95% CI: 1.6 mm, 3.5 mm) [p = 0.0001].
Days of disability
Compared to placebo, Aclasta significantly reduced the average days of reduced activity and days of bed rest due to low back pain by 17.9 days and 11.3 days, respectively, while also reducing the average days of reduced activity. and bed rest days due to fractures of 2.9 days and 0.5 days, respectively, compared with placebo (p = 0.01).
Clinical efficacy in the treatment of osteoporosis in patients at increased risk of fractures after a recent hip fracture (RFT)
The incidence of clinical, vertebral, non-vertebral fractures and hip fractures included was evaluated in 2,127 men and women aged 50-95 years (mean age 74.5 years) with a recent (within 90 days) hip fracture due to mild trauma that had been followed with study treatment (Aclasta) for an average of 2 years. In approximately 42% of patients the femoral neck T-score was less than -2.5 and in approximately 45% of patients had a femoral neck T-score greater than -2.5. Aclasta was administered annually until clinical fractures were confirmed in at least 211 patients of the study population. Vitamin D levels were not routinely measured but a loading dose of vitamin D (50,000 to 125,000 IU orally or intramuscularly) was given to the majority of patients 2 weeks prior to infusion. All participants had taken 1,000 to 1,500 mg of elemental calcium plus 800 to 1,200 IU of vitamin D supplement daily. 95% of patients received the infusion two or more weeks after hip fracture repair and median time to The infusion was approximately six weeks after hip fracture repair. The primary efficacy variable was the incidence of clinical fractures throughout the study.
Effect on all clinical fractures
The incidence rates of the main clinical fracture variables are presented in Table 4.
Table 4 Comparison between treatments in the incidence of the main clinical fracture variables
The study was not designed to measure significant differences in hip fracture but a trend in favor of reducing new hip fractures was observed. In the Aclasta treatment group, all cause mortality was 10% (101 patients) compared to 13% (141 patients) in the placebo group. This corresponds to a reduction in the risk of all-cause mortality of 28% (p = 0.01).
The incidence of delayed hip fracture healing was comparable between Aclasta (34 [3.2%]) and placebo (29 [2.7%]).
Effect on bone mineral density (BMD)
In the HORIZON-RFT study, Aclasta treatment significantly increased total hip and femoral neck BMD relative to placebo treatment across all timepoints. Aclasta treatment showed an increase of 5.4%. total hip BMD and 4.3% femoral neck over 24 months of treatment compared to placebo.
Clinical efficacy in humans
In the HORIZON-RFT study, 508 men were randomized and 185 patients were evaluated for BMD at Month 24. A similar significant 3.6% increase in comparable total hip BMD was observed at Month 24 in patients treated with Aclasta. the effects observed in postmenopausal women in the HORIZON-PFT study. The study was not sized to demonstrate a reduction in clinical fractures in humans; the incidence of clinical fractures was 7.5% in men treated with Aclasta compared with 8.7% in placebo. "male (study CZOL446M & SUP2; 308) the percentage change in spine BMD at month 24 relative to baseline was non-lower after an annual" infusion of Aclasta compared to alendronate given weekly.
Clinical efficacy in osteoporosis induced by long-term systemic glucocorticoid therapy The efficacy and safety of Aclasta in the treatment and prevention of osteoporosis induced by long-term systemic glucocorticoid therapy were evaluated in a randomized, multicentre study in double-blind, stratified, with active control on 833 men and women aged 18-85 years (mean age for men 56.4 years; for women 53.5 years) treated with> 7.5 mg / day of prednisone orally (or equivalent). Patients were stratified by duration of glucocorticoid treatment prior to randomization (≤3 months versus> 3 months). Study duration was one year. Patients were randomized to Aclasta 5 mg single infusion or oral risedronate 5 mg daily for one year. All had received 1,000 mg of elemental calcium daily plus 400 to 1,000 IU vitamin D supplementation. cia was demonstrated in a non-inferiority design to risedronate by sequentially showing the percent change in spine BMD at month 12 from baseline in the treatment and prevention subpopulations, respectively. The majority of patients continued taking glucocorticoids for the one year duration of the study.
Effect on bone mineral density (BMD)
The increases in BMD in the spine and femoral neck at month 12 were significantly greater in the Aclasta treatment group compared to risedronate (p
Clinical efficacy in the treatment of Paget's disease of bone Aclasta has been studied in male and female patients over 30 years of age with mainly mild to moderate Paget's disease of bone (mean serum alkaline phosphatase level 2, 6-3.0 times the age-specific upper limit of normal at study enrollment) confirmed by radiological examination.
The efficacy of a 5 mg infusion of zoledronic acid versus risedronate 30 mg daily administered for 2 months was demonstrated in two comparator studies of 6 months duration. After 6 months, Aclasta showed rates of 96% (169/176) and 89% (156/176) of therapeutic response and normalization of serum alkaline phosphatase (SAP) compared to 74% (127/171) and 58% ( 99/171) obtained with risedronate (always p
With the pooled results, a similar decrease in pain severity and pain interference scores over 6 months from baseline was seen for Aclasta and risedronate.
Patients who were classified as responders to treatment at the end of the 6-month baseline study were considered eligible to be included in the extended evaluation period. Of the 153 patients treated with Aclasta and 115 patients treated with risedronate who entered the extended observation period of the study, after a mean follow-up period of 3.8 years after administration, the proportion of patients who completed the study prolonged observation due to the need for retreatment (clinical judgment) was greater for risedronate (48 patients, 41.7%) than for zoledronic acid (11 patients, 7.2%). The mean time to termination of the prolonged observation period due to the need for retreatment of Paget from the initial dose was longer for zoledronic acid (7.7 years) than for risedronate (5.1 years).
Six patients who achieved a therapeutic response 6 months after treatment with Aclasta and who then had disease recurrence during the prolonged evaluation period were retreated with Aclasta after a mean time of 6.5 years between initial and the reprocessing. Five of the 6 patients had serum alkaline phosphatase levels within the normal range at month 6 (Last Observation Carried Forward, LOCF).
Bone histology was evaluated in 7 patients with Paget's disease 6 months after treatment with 5 mg zoledronic acid. Bone biopsy results showed normal bone quality with no evidence of impaired bone remodeling and without evidence of mineralization defects. These results were in agreement with the biochemical marker of evidence of normalization of bone turnover.
The European Medicines Agency has waived the obligation to submit the results of studies with Aclasta in all subsets of the pediatric population for Paget's disease of the bone, osteoporosis in postmenopausal women at increased risk of fracture , osteoporosis in men at increased risk of fracture and the prevention of clinical fractures after a hip fracture in men and women (see section 4.2 for information on pediatric use).
05.2 Pharmacokinetic properties
Single and multiple 5 and 15 minute infusions of 2, 4, 8 and 16 mg zoledronic acid in 64 patients showed the following pharmacokinetic data, regardless of dose.
Distribution
After the start of the zoledronic acid infusion, plasma concentrations of the active substance increased rapidly, peaking at the end of the infusion period, followed by a rapid decrease.
Elimination
After intravenous administration, zoledronic acid is eliminated by a three-step process: rapid disappearance with a biphasic course from the systemic circulation, with half-lives of t½a 0.24 and t½b 1.87 hours, followed by a long elimination phase with terminal elimination half-life of t½g 146 hours. No accumulation of the active substance was observed in plasma after multiple doses administered every 28 days. bone uptake and renal excretion. Zoledronic acid is not metabolised and is excreted unchanged via the kidney. During the first 24 hours, 39 ± 16% of the administered dose is recovered in the urine, while the remainder is mainly bound to bone tissue. This absorption into bone is common for all bisphosphonates and is presumably a consequence of the structural analogy to pyrophosphate. As with other bisphosphonates, the retention time of zoledronic acid in the bones is very long. From the bone the drug is very slowly released into the systemic circulation and then eliminated via the kidney. Total body clearance is 5.04 ± 2.5 l / h, regardless of dose, and is not influenced by gender, age, race or body weight. The variation in plasma clearance of zoledronic acid between and within individuals was 36% and 34%, respectively. Increasing the infusion time from 5 to 15 minutes resulted in a 30% decrease in zoledronic acid concentration at the end of the infusion, but had no effect on the area under the plasma concentration versus time curve.
Pharmacokinetic / pharmacodynamic relationships
No interaction studies have been performed with other medicinal products and zoledronic acid. Since zoledronic acid is not metabolised in humans and the substance has been found to have little or no capacity as a direct-acting and / or irreversible metabolism inhibitor. dependent on P450 enzymes, zoledronic acid is unlikely to reduce the metabolic clearance of substances metabolised via cytochrome P450 enzyme systems. Zoledronic acid is not extensively bound to plasma proteins (approximately 43-55% bound ) and the bond is independent of the concentration. Therefore, interactions resulting from the displacement of highly protein-bound medicinal products are unlikely.
Special populations (see section 4.2)
Renal impairment
Renal clearance of zoledronic acid was correlated with creatinine clearance, since renal clearance accounts for 75 ± 33% of creatinine clearance, which averaged 84 ± 29 ml / min in the 64 patients studied. (range 22 to 143 mL / min). The small increases observed in AUC (0-24hr), between approximately 30% and 40% in mild to moderate renal impairment, compared to patients with normal renal function, and the absence of drug accumulation following multiple doses irrespective of renal function, suggest that no dose adjustments of zoledronic acid are required in mild (Clcr = 50-80 ml / min) and moderate renal impairment up to creatinine clearance of 35 ml / min. The use of Aclasta in patients with severe renal impairment (creatinine clearance
05.3 Preclinical safety data
Acute toxicity
The maximum non-lethal dose for single intravenous administration was 10 mg / kg body weight in the mouse and 0.6 mg / kg in the rat. In dog single dose infusion studies, 1.0 mg / kg (6 times the recommended human therapeutic exposure based on AUC) administered over 15 minutes was well tolerated with no renal effects.
Subchronic and chronic toxicity
In intravenous infusion studies, renal tolerability of zoledronic acid was established in rats with administration of 0.6 mg / kg as 15-minute infusions at 3-day intervals, for a total of six infusions (for a cumulative dose which corresponds to AUC levels of approximately 6 times the therapeutic exposure in humans) while five 15-minute infusions of 0.25 mg / kg administered at 2-3 week intervals (a cumulative dose corresponding to 7 times l " human therapeutic exposure) were well tolerated in dogs. In intravenous bolus studies, doses that were well tolerated decreased with increasing study duration: doses of 0.2 and 0.02 mg / kg per day were well tolerated for 4 weeks in rats and dogs, respectively, but only doses of 0.01 mg / kg and 0.005 mg / kg were well tolerated in rats and dogs, respectively, when administered for 52 weeks.
Long-term repeated administration, at cumulative exposures sufficiently in excess of the maximum expected human exposure, produced toxicological effects in other organs, including the gastrointestinal tract and liver, and at the site of intravenous administration. The clinical relevance of these findings is unknown. The most frequent finding in repeat-dose studies is an increase in spongy bone tissue in the metaphyses of long bones in developing animals at nearly all doses, reflecting the product's anti-resorptive pharmacological activity.
Reproductive toxicity
Teratology studies were performed in two species, both employing subcutaneous administration. Teratogenicity was observed in rats at doses ≥0.2 mg / kg and resulted in external, visceral and skeletal malformations. Dystocia was observed at the lowest dose tested in the rat (0.01 mg / kg body weight). No teratogenic or embryo / fetal effects were observed in rabbits, although maternal toxicity was marked at the dose of 0.1 mg / kg due to low serum calcium levels.
Mutagenicity and carcinogenic potential
Zoledronic acid was not mutagenic in the mutagenicity tests performed and the carcinogenicity tests did not provide evidence of carcinogenic potential.
06.0 PHARMACEUTICAL INFORMATION
06.1 Excipients
Mannitol
Sodium citrate
Water for injections
06.2 Incompatibility
This medicinal product should not come into contact with calcium-containing solutions. Aclasta must not be mixed or administered intravenously with other medicinal products.
06.3 Period of validity
Unopened bottle: 3 years
After opening: 24 hours at 2 ° C - 8 ° C
From a microbiological point of view, the product should be used immediately. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user and would normally not be longer than 24 hours at 2 ° C - 8 ° C.
06.4 Special precautions for storage
This medicine does not require any special storage conditions.
For storage conditions of the medicinal product after first opening, see section 6.3.
06.5 Nature of the immediate packaging and contents of the package
100 ml of solution in a clear plastic bottle (cycloolefin polymer) closed with a fluoropolymer coated bromobutyl rubber stopper and aluminum / polypropylene cap with flip off element.
Aclasta is supplied in single packs containing one bottle or in multipacks consisting of five packs, each containing one bottle.
Not all pack sizes may be marketed.
06.6 Instructions for use and handling
For single use only.
The solution should only be used if it is clear, free from particles or discolouration.
If stored in the refrigerator, allow the solution to reach room temperature before administration. Aseptic techniques must be followed during the preparation of the infusion. The unused medicinal product and the waste derived from this medicinal product must be disposed of in accordance with local regulations.
07.0 MARKETING AUTHORIZATION HOLDER
Novartis Europharm Limited
Frimley Business Park
Camberley GU16 7SR
UK
08.0 MARKETING AUTHORIZATION NUMBER
EU / 1/05/308/001
EU / 1/05/308/002
037105018
09.0 DATE OF FIRST AUTHORIZATION OR RENEWAL OF THE AUTHORIZATION
Date of first authorization: 15 April 2005
Last renewal date: April 19, 2015








.jpg)